










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkAnales de la Facultad de Medicina
Print version ISSN 1025-5583
An. Fac. med. vol.69 no.4 Lima Dec. 2008
Síndrome de Woodhouse Sakati
Woodhouse-Sakati syndrome: a case report
Franklin Aranda1,2, Miguel Chávez3, Angel Quispe-Mauricio4, Adolfo Caro1,2
1 Médico Pediatra, Instituto Nacional de Salud del Niño (INSN). Lima, Perú.
2 Profesor invitado, Facultad de Medicina de la Universidad Nacional Mayor de San Marcos. Lima, Perú.
3 Médico Genetista, Servicio de Genética, Instituto Nacional de Salud del Niño. Lima, Perú
4 Médico Cirujano, Red Asistencial Pasco, Seguro Social de Salud-EsSalud. Pasco, Perú.
Resumen
Presentamos un caso de síndrome de Woodhouse Sakati, en una paciente de 11 años de edad, quien presentó alopecia congénita, hipoacusia neurosensorial bilateral, diabetes mellitus insulino dependiente, hipogonadismo primario, retardo del desarrollo psicomotor, comunicación interventricular y disminución de somatomedina C (IGF1). La evolución y el tratamiento de soporte fueron satisfactorios.
Palabras clave: Woodhouse Sakati; receptor IGF tipo 1; alopecia; diabetes mellitus; hipogonadismo.
Abstract
We present a rare case of Woodhouse-Sakati syndrome in an 11 year-old patient, who presented congenital alopecia, bilateral sensorineural hearing loss, insulin-dependent diabetes mellitus, primary hypogonadism, psychomotor retardation, interventricular communication, decreased IGF1. The evolution and supportive treatment were satisfactory.
Key words: Woodhouse-Sakati syndrome; receptor, IGF type 1; alopecia; diabetes mellitus; hypogonadism.
INTRODUCCIÓN
El síndrome de Woodhouse Sakati es una enfermedad extremadamente rara, genéticamente determinada, de tipo autosómica recesiva, que fue descrita por primera vez en seis pacientes de dos familias en Arabia Saudita (1); posteriormente, el síndrome fue comunicado en un hombre de Turquía, con retardo mental, alopecia parcial, diabetes mellitus, hipogonadismo y sordera (2).
El año 2007 (3), se publicó sobre doce familias con un desorden multisistémico autosómico recesivo, que había sido descrito previamente en una comunicación de casos, en el año 1996 (4). En esta, diez de las familias fueron consanguíneas y una ya había sido descrita previamente por Woodhouse y Sakati (1), en el año 1983.
Las manifestaciones clínicas de esta enfermedad han sido variadas, desde características muy frecuentes -como alopecia, sordera neurosensorial, retardo mental en grado variable, ausencia de cejas, alteraciones electrocardiográficas, diabetes mellitus insulino dependiente, hipogonadismo hipo e hipergonadotrópico, retardo puberal-, hasta características menos frecuentes, como retraso en el desarrollo psicomotor, disartria, características dismórficas (frente amplia, occipucio plano, cara triangular, nariz prominente, hipertelorismo y fisura palpebral baja), hiperreflexia, síndrome neurológico extrapiramidal con distonia y movimientos coreoatetósicos, cambios cerebrales en la resonancia magnética nuclear y, en algunos casos, con manifestaciones oculares, como keratoconus (5).
CASO CLÍNICO
Paciente de 11 años y 3 meses de edad, natural y procedente de Villa El Salvador, Lima-Perú, quien fue traída por su abuela materna a emergencia del Instituto Nacional de Salud del Niño (INSN), con diagnóstico previo hacía tres años de diabetes mellitus tipo 1, en tratamiento irregular. La abuela refiere que desde hacía un mes la paciente presentó poliuria, polidipsia y polifagia. Una semana antes de su ingreso, tuvo tos con expectoración blanquecina y que al examen médico se evidenció faringe congestiva, con dificultad respiratoria leve, por lo que fue tratada con amoxicilina 250 mg, vía oral, cada ocho horas, por siete días, remitiendo el proceso respiratorio. Cinco días antes de su ingreso presentó náuseas, vómitos posprandiales y dolor abdominal en epigastrio, que cedieron con antiespasmódicos. Además, hubo deposiciones semilíquidas, sin moco, sangre, pujo ni tenesmo, por lo que acudió a un centro de salud, donde al comprobar glucosa sérica en 500 mg/dL decidieron derivarla a nuestro hospital, para manejo especializado.
La paciente era producto de una primera gestación a término, sin controles prenatales, parto vaginal eutócico, 2 200 gramos al nacer, sin asfixia perinatal, lactancia materna exclusiva hasta los siete meses de edad, inmunizaciones completas para el esquema de vacunación peruano. En su tarjeta de crecimiento y desarrollo se señaló retardo del desarrollo psicomotor. Había sido operada de cierre de comunicación interventricular (CIV) y de banding de arteria pulmonar a los dos años de edad. Se negó antecedentes familiares de diabetes. Los padres eran peruanos, y la bisabuela por línea materna de origen extranjero, desconocido.
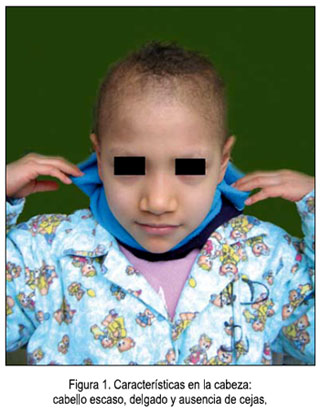
Al examen físico, el peso fue 20 kg, la talla 1,15 m, frecuencia cardiaca 86 lpm, frecuencia respiratoria 22 rpm, temperatura 36,5°C, la paciente siempre relacionada con el entorno. En la piel, se evidenció palidez marcada, sin cianosis y un leve edema pretibial. En la cabeza, llamó nuestra atención el cabello escaso, delgado y la ausencia de cejas (figura 1); la información dada por la abuela refirió que esa característica la conservaba desde el nacimiento. En los ojos, conducto auditivo externo y boca no se encontraron alteraciones, salvo caries dentales. No se palparon adenopatías ni visceromegalia. En la evaluación pulmonar, no hubo hallazgo patológico y, en el cardiovascular, un soplo sistólico multifocal II/VI. Comprobamos puño percusión lumbar derecha positiva y puntos renoureterales negativos. Había discreto edema vulvar, con secreción blanquecina sin mal olor. No se evidenció déficit motor o sensorial ni reflejos patológicos. Colaboradora con el examen, presentó buena empatía con los examinadores y con el personal de salud del servicio de hospitalización. Llamó la atención desde un inicio su voz delgada, definida como aflautada.
La paciente tuvo diagnóstico de diabetes mellitus descompensada y se inició tratamiento en emergencia con hidratación, con suero fisiológico, y protocolo de manejo con insulina, para dicha patología. Una vez compensada, se reinició la vía oral y se realizó el manejo con dieta, insulina y controles regulares de glicemia. Fue durante su hospitalización donde se sospechó del síndrome de Woodhouse Sakati.
Entre los exámenes auxiliares, la hemoglobina fue 11,2 mg/dL, hematocrito 38%, plaquetas 400 000/mL, leucocitos 6 300/mL, eosinófilos 2%, abastonados 0%, segmentados 71%, linfocitos 21%, monocitos 6%. En el frotís de sangre periférica había hipocromía 2+, anisocitosis 1+. La glucosa sérica fue 426,7 mg/dL, hemoglobina glicosilada 35,9%, urea 27,9 mg/dL, creatinina 0,50 mg/dL, proteínas totales 7,8g/dL, albúmina 4,4g/dL, globulina 3,4g/dL, colesterol 204,3mg/dL; entre los electrolitos séricos, sodio 133,00 mmol/L, potasio 3,41 mmol/L, cloro 107,00 mmol/L; microalbuminuria 11,5 mg/24 horas. Tiempo de coagulación 630, tiempo de sangría 200, tiempo de protrombina 13,3, tiempo de tromboplastina parcial activada 39,3. TSH ultrasensible 2,09 mIU/dL, T4 libre 100 nmol/L, hormona luteinizante 0,62 mIU/mL, hormona folículo estimulante 6,9 mIU/mL, estradiol 146 pmol/L, somatomedina C (IGF1) 97 ng/mL, prueba de Elisa para VIH negativo. En el cultivo de secreción vaginal, leucocitos 15 a 20/campo, hematíes 0 a 1 /campo. A la coloración Gram, escasos cocos Gram positivos y bacilos Gram negativos; no había células clave o levaduras. El examen parasitológico fue negativo para tricomonas; cultivo negativo a gérmenes patógenos.
Por informe radiológico, la edad ósea según TW-2 fue 9,2 años.
El informe ecocardiográfico señaló paciente postoperada de banding + comunicación interventricular (CIV); CIV residual de 4 mm, estenosis supra valvular (de banding) 30 mmHg, insuficiencia mitral leve, crecimiento de cámaras izquierdas de grado leve, función de ventrículo izquierdo normal.
En la exploración audiológica, se halló hipoacusia bilateral moderada a severa (85 000 Hz), neurosensorial.
En la ecografía abdominal, el hígado medía 13 cm de longitud, con aumento difuso de ecogenicidad, vesícula y vías biliares normales, páncreas normal, bazo de 8,3 cm de longitud de aspecto normal, no ascitis, asas intestinales hiperperistálticas, con bastante contenido líquido. En la ecografía renal, el riñón derecho medía 9,3 cm de longitud, anteroposterior 3,9 cm, parénquima desdiferenciado de 1,7 cm de longitud, pelvis de 7 cm x 3,2 de diámetro anteroposterior, uréter proximal medía 6,3 mm (premiccional), mínimo colapso de la pelvis en el control posmiccional. El riñón izquierdo de 10,8 cm de longitud, AP= 4,2 cm, parénquima desdiferenciado de 1,5 cm, pelvis renal (posmiccional) L=6,6 , AP=5,3 cm, con presencia de ecos internos, urotelio de 3 mm y un uréter distal de 7 mm, vejiga con abundante residuo, de paredes normales.
La evaluación psicológica encontró coeficiente intelectual total 74, categoría fronterizo, coeficiente intelectual potencial de 90, categoría normal promedio, edad mental de 8 años 6 meses.
En la evaluación dermatológica, pelo levemente ensortijado, ralo, de mala implantación; el estudio determinó alopecia congénita. En la evaluación odontopediátrica, caries dental en 11 pares dentales. Evaluación ginecológica, vulvovaginitis; se indica aseo vulvar dos veces al día y luego de deposiciones. Evaluación endocrinológica, paciente con diabetes mellitus descompensada más hipogonadismo primario. Evaluación urológica, vejiga retencionista, que no muestra reflujo durante la micción. Escaso residuo posmiccional (figura 2). En la evaluación cardiológica, había hipertensión pulmonar leve. Evaluación oftalmológica, catarata de ambos ojos, glaucoma congénito tardío. Y en la evaluación genética, se sospechó síndrome de Woodhouse Sakati.
DISCUSIÓN
La tasa alta de consanguinidad entre la población árabe -como en Turquía y específicamente en Arabia Saudita- permite una incidencia alta de serias enfermedades genéticas de tipo recesiva, como el síndrome de Woodhouse Sakati. Es así que nosotros creemos que el antecedente familiar de procedencia árabe es muy importante (el apellido Amezquita de la línea materna tiene origen en la invasión árabe a España).
Se encontró alopecia parcial progresiva e hipogonadismo, además de ausencia de vello axilar y púbico, en nuestro caso. El hipogonadismo fue diagnosticado con criterio clínico y de laboratorio; esto se corrobora con los hallazgos de cambios histopatológicos, mostrados por Woodhouse and Sakati (1983) (1).
Nuestro caso presentó diabetes mellitus insulino dependiente, sin asociación a hipercolesterolemia.
No se halló asociación a desorden progresivo extrapiramidal, lo cual ha sido comunicado por Devriendt (4); sin embargo, para Al-Semari (3) no es un hallazgo consistente. Las comunicaciones de casos del síndrome muestran que el comienzo de las manifestaciones neurológicas son entre los 10 y 20 años de edad. Nuestra paciente ya tenía 11 años y aún no mostraba clínica de dichas manifestaciones; sin embargo, en la evaluación neuropsicológica mostró un coeficiente intelectual total 74, que la ubicaba en la categoría de fronteriza.
Llamó nuestra atención que en la mayoría de pacientes encontrados en la literatura, así como en nuestra paciente, exista una franca disminución del IGF1 (insulin like growth factor-1), lo que hace pensar que probablemente podría ser un nuevo tipo de síndrome neuro-endocrino-ectodermo, como lo planteó Al-Semari and Bohlega (3).
Se ha reconocido como componentes de este síndrome la sordera neurosensorial, diabetes mellitus tipo 1, intolerancia a la glucosa, disminución del IGF1 y alteraciones electrocardiográficas (producto de algunas malformaciones cardiacas), como lo descrito en el presente caso (1).
REFERENCIAS BIBLIOGRÁFICAS
1. Woodhouse NJ, Sakati NA. A syndrome of hypogonadisim, alopecia, diabetes mellitus, mental retardation, deafness and ECG abnormalities. J Med Genet. 1983;20:216-19. [ Links ]
2. Gul D, Ozata M, Mergen H, Odabazi Z, Mergen M. Woodhouse and Sakati syndrome: report of a new patient. Clin Dysmorph. 2000;9:123-5. [ Links ]
3. Al-Semari A, Bohlega S. Autosomal-recessive syndrome with alopecia, hypogonadism, progressive extra-pyramidaldisorder, white matter disease, sensory neural deafness, diabetes mellitus, and low IGF1. Am J Med Genet A. 2007;143(2):149-60. [ Links ]
4. Devriendt K, Legius E, Fryns J. Progressive extrapyramidal disorder with primary hypogonadism and alopecia in sibs: a new syndrome? Am J Med Genet. 1996;62:54-7. [ Links ]
5. Al-Swailem SA, Al-Assiri AA, Al-Torbak AA. Woodhouse Sakati syndrome associated with bilateral keratoconus. Br J Ophthalmol.2006;90:116-7. [ Links ]
Manuscrito recibido el 2 de diciembre de 2008 y aceptado para publicación el 22 de diciembre de 2008.
Correspondencia:
Dr. Franklin Aranda Paniora
Av. Brasil 600, Breña
Lima 5, Perú
Correo-e: faranda@hotmail.com

