










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. v.70 n.3 Lima sep. 2009
Neurofibromatosis tipo 1 asociada a déficit cognitivo, hidrocefalia, displasia ósea y elefantiasis
Neurofibromatosis type 1 associated to cognition deficit, hydrocephaly, bone dysplasia and elephantiasis
Julio Ramírez1,2, Gustavo Wong3, Victor Mechán1,4, Cesar Durán5, Gustavo Cerrillo6, Gerardo Ronceros7,8, Isabel Ballena9, Miguel Llanos10
1 Docente de la Facultad de Medicina, Universidad Nacional Mayor de San Marcos. Lima, Perú.
2 Servicio de Medicina Interna, Hospital Nacional Dos de Mayo. Lima, Perú.
3 Servicio de Neurología, Hospital Nacional Dos de Mayo. Lima, Perú.
4 Servicio de Hematología, Hospital Nacional Dos de Mayo. Lima, Perú.
5 Servicio de Oftalmología, Hospital Nacional Dos de Mayo. Lima, Perú.
6 Servicio de Anatomía Patológica, Hospital Nacional Dos de Mayo. Lima, Perú.
7 Departamento de Patología Clínica, Hospital Nacional Dos de Mayo. Lima, Perú.
8 Instituto de Investigaciones Clinicas, Facultad de Medicina, Universidad Nacional Mayor de San Marcos. Lima, Perú.
9 Servicio de Radiología, Hospital Nacional Dos de Mayo. Lima, Perú.
10 Alumno del 5° Año de la Escuela Académico Profesional de Medicina, Facultad de Medicina, Universidad Nacional Mayor de San Marcos. Lima, Perú.
Resumen
Se presenta el caso de un paciente varón de 31 años, procedente de Cajamarca, con problemas cognitivos desde su escolaridad temprana, que desde los 12 años desarrolló incremento progresivo de volumen y deformación de su miembro inferior derecho y en menor cuantía del izquierdo, asociado a plegamiento de piel, crecimiento de nódulos subcutáneos, con formación de cordones nerviosos en muslo y pierna que le daban un aspecto elefantiásico. Dolor y limitación funcional para la marcha. Concomitantemente, presencia en piel de manchas café con leche generalizadas y nódulos de Lisch en ambos iris. En la resonancia magnética y TAC con contraste, se evidenciaron abundantes nódulos plexiformes en cavidad abdominopélvica, que continuaban por todo el muslo, de tamaño uniforme, formando tumoraciones y cordones. Asimismo, presentó displasia ósea (adelgazamiento de la cortical del fémur y tibia, formación de neocótilo izquierdo, pseudoartrosis). La resonancia cerebral con contraste descubrió una hidrocefalia triventricular, normotensa.
Palabras clave: Neurofibromatosis 1; hidrocefalia; trastornos cognitivos; displasia ósea; elefantiasis.
Abstract
We report the case of a 31 year-old male from Cajamarca, Peru, with cognitive problems since childhood, who at 12 year-old developed increased volume and deformation of his right lower limb and to a lesser extent of his left, associated to massive skin folding, growth of subcutaneous nodules, elephantine look due to nerve cords formation in thigh and leg, pain and functional walking limitation. Concomitantly generalized skin café au lait spots, subcutaneous nodules, and Lisch nodules in both irides. Magnetic resonance and contrast TAC revealed abundant uniform plexiform nodules in abdomino-pelvic cavity forming masses and cords that continued throughout his thigh as lumps and cords. In addition, the patient presented bone dysplasia (thinning of femur and tibia cortex, pseudoarthrosis, new left cotyle formation). Contrast magnetic resonance showed triventricular normotensive hydrocephalus.
Key words: Neurofibromatosis 1; hydrocephalus; cognition disorders; bone dysplasia; elephantiasis.
INTRODUCCIÓN
La neurofibromatosis tipo 1 (NF1) es un desorden neurocutáneo autosómico dominante, con una incidencia al nacer de 1/2 500 y prevalencia mínima de 1/4 000 - 5 000 personas (1), siendo heredada en 50% de casos, mientras el porcentaje restante es explicado por mutación espontánea. Son criterios diagnósticos para esta enfermedad las manchas café con leche, neurofibromas plexiformes, pecas en axila o ingle, nódulos de Lisch (hamartomas pigmentarios del iris), gliomas de la vía óptica, displasia de huesos largos o ala esfenoidal y un familiar en primer grado con NF1. Algunos pacientes presentan deterioro cognitivo, consistente en coeficiente intelectual por debajo del promedio, problemas de conducta y aprendizaje (2).
Aunque en el Perú no existen cifras oficiales de su incidencia, un estudio realizado en el servicio de Pediatría de un Hospital de Lima (1989-1998), objetivó 2 914 atenciones, correspondiendo 47 casos (1,6%) a síndromes neuroectodérmicos. De estos, 21 (44,4%) correspondieron a neurofibromatosis (3). El gen NF1 se ubica en el cromosoma 17 (q11.2), siendo su producto la neurofibromina, un potente reductor de la proliferación celular, que promueve la inactivación del p21ras (4).
Motiva la presentación de este caso, reportar y analizar 4 complicaciones de NF1: a) déficit cognitivo; b) elefantiasis neurofibromatosa, rara anomalía caracterizada por crecimiento exagerado de neurofibromas a lo largo de los nervios periféricos; c) hidrocefalia triventricular, complicación neurológica no tan rara, a juzgar por los resultados de autopsias; y, d) displasias óseas.
CASO CLÍNICO
Paciente varón, mestizo, de 31 años, natural y procedente de la provincia de San Miguel, del departamento de Cajamarca. Durante su escolaridad temprana no aprendió a leer ni a escribir, por lo que fue retirado de la escuela al reprobar en dos oportunidades el primer grado. Durante su infancia no dedicó atención a los estudios, solo al juego. Refieren sus familiares que desde su pubertad percibieron incremento de volumen del muslo derecho, con progresivo avance distal, debilidad y deformidad del miembro inferior derecho, que le imposibilitó paulatinamente la deambulación. Refieren además sueño disminuido, sudoración incrementada. Orina cada 3 horas, levantándose por la noche para miccionar 2 a 3 veces.
Desde hace 12 años, percibieron múltiples nódulos subcutáneos en su cuerpo. Hace 8 años se incrementó la limitación funcional del miembro inferior derecho, asociándose a dolor de cadera, por lo que acudió durante dos años a un centro de salud primario de San Miguel de Cajamarca, donde le prescribieron dexametasona 4 mg/IM/1 vez/semana y ® dexacort oral 1 tableta diaria, para calmar su dolor de cadera, hasta hace 1 año. Desde hace 9 meses yacía postrado en cama, por dolor de cadera y limitación funcional, a causa del peso del miembro afectado. Vivienda de adobe, sin servicios básicos, con dos habitaciones, donde habitaban 7 personas. Nacido de parto a término, caminó y habló al año y medio. A los 8 años, estuvo inmovilizado 4 meses por dolor en rodilla derecha. Erección y eyaculación, tras masturbarse; no refería hijos. Bebedor social, consumía café todos los días. Padre de 65 y madre de 55 años, sin manchas café con leche. Tenía 7 hermanos, de los cuales un hermano varón de 33 años y sus hermanas mujeres de 27 y 24 años presentaban de 2 a 4 manchas café con leche en abdomen y espalda.
Al examen físico, aparecía lúcido, orientado en tiempo y espacio, regular estado general, regular estado de nutrición e hidratación; presión arterial 120/70; peso 66 kg; talla 1,45 m (apoyado en pie izquierdo; vértex-pubis 0,67 m; pubis-ras del pie izquierdo 0,73 m; pubis-ras del pie derecho 0,8 m). Distancia espina ilíaca-maléolo interno 0,87 m y 0,67 m, en miembro inferior derecho y miembro inferior izquierdo, respectivamente. Circunferencia de muslo 0,92 m y 0,45 m en miembro inferior izquierdo y miembro inferior derecho, respectivamente. En miembro inferior derecho mostraba notable incremento de volumen, piel seca, hipertrófica, color café, donde se palpaba múltiples nódulos subcutáneos indoloros y tumoraciones pétreas, inmóviles, de bordes regulares (15x7 y 25x10 cm), a predominio de muslo y talón. Aunque la pierna no presentaba muchos nódulos, exhibía plegamientos de piel y tejido conectivo que limitaban la marcha, flexión y rotación de rodilla, manteniendo al miembro inferior derecho en permanente rotación externa. Manchas café con leche de 5 a 7 cm en abdomen, en miembro inferior izquierdo y dorso. Múltiples nódulos cutáneos y subcutáneos en todo el cuerpo. Articulaciones hiperextendidas. Escoliosis lumbar.
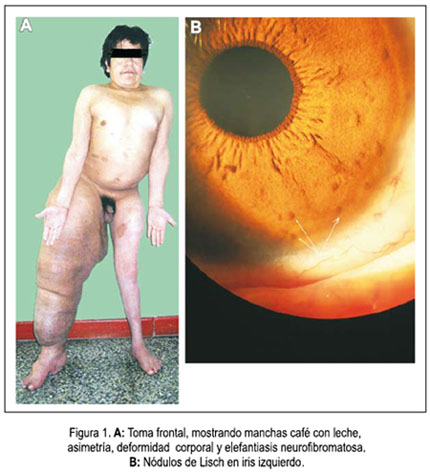
El cráneo era normocéfalo, con perímetro cefálico de 56 cm. Expansión torácica en reposo y en expansión máxima 94,3 cm y 95,5 cm, respectivamente. Cardiovascular, pulmones y abdomen no fueron contributorios. Testículos 3,3 x 1,8 y 3 x 2 cm, derecho e izquierdo, respectivamente. Vello pubiano triangular, escaso, con base superior. Al examen neurológico, hipoactivo, no prestaba adecuada atención. No ejecutaba una de 3 órdenes, olvidaba 2 de 3 palabras a repetir, al minuto y a los 5 minutos. Aunque repetía de manera ordenada y en reversa los días de la semana, no lograba imitar un cubo o pentágonos entrelazados, delineándolos solo con trazos bidimensionales. Tras 5 intentos, era incapaz de repetir una frase. Facies discretamente asimétrica, con leve ptosis del ojo izquierdo. Hipotrofia de músculos temporales y maseteros. Escuchaba voz susurrada hasta 50 cm de distancia. Rinné positivo, aunque la conducción aérea no llegaba a ser el doble de la ósea. Hemilengua derecha protruida, incrementada de volumen. Neurofibromas plexiformes (8 y 2 cm de diámetro) en región retroauricular derecha y submaxilar izquierda. Fuerza muscular en miembros superiores 4+/5+, simétrica. Paraparesia derecha, distal 0+/5+ y 3+/5+ en miembro inferior izquierdo. Arreflexia osteotendinosa generalizada. Reflejos cutaneoabdominal y cremasteriano ausentes. Ausencia de reflejo rotuliano, aquíleo y medio plantar en miembro inferior derecho. Babinski indiferente en miembro inferior derecho. Hipotonía discreta en los cuatro miembros. Sensibilidad al tacto disminuida distalmente, a predominio de dedos de pies, más notable en el lado derecho. Banda hiperalgésica D8 a D12 y en dermatomas S3–S4–S5 y coccígeo, con leve asimetría a predominio derecho. Discriminación de dos puntos deficiente en miembros inferiores, más marcada en pie derecho. Hipopalestesia distal en miembros inferiores, a predominio distal derecho. Prueba índice-nariz con temblor, a predominio izquierdo. Torpeza motora, disritmocinesia y disdiadococinesia bilateral, a predominio izquierdo. figuras 1, 2 y 3.
Los exámenes auxiliares realizados el 5/04/2009 mostraron glucosa 69 mg/100 mL, fibrinógeno 525 mg/dL (N: 200-400), cortisol matutino 2,26 µg/mL (N: 7-28); hemoglobina 14,7 g/dL, leucocitos 7 600/mL, plaquetas 391 000/mL, volumen corpuscular medio 86,5 fL (fentolitros), hemoglobina corpuscular media 28,8 µL, eosinófilos 6, abastonados 2, segmentados 62, linfocitos 24, monocitos 6; grupo sanguíneo A1, factor RH(+); creatinina 0,6 mg/dL; urea 28 mg/dL (N: 20-40). El día 28/4/2009, proteínas totales 6,1 g/dL; albúmina 3,4 g/dL; heces con presencia de Trichomonas hominis; TSH 7,9 uUI/mL (N: 0,35-5,5); FSH 7,71 uUI (N: 1,4-18); LH 6,8 uUI/mL (N: 1,7-8,6); testoterona 264,7 ng/dL (N: 241-827); sodio 133 meq/L, potasio 3,9 meq/L, cloro 114 meq/L; anticuerpos IgG HIV-1 y 2 no reactivos; RPR no reactivo; HBsAg no reactivo; tiempo de protrombina 14,5, tiempo parcial de protrombina 38,8.
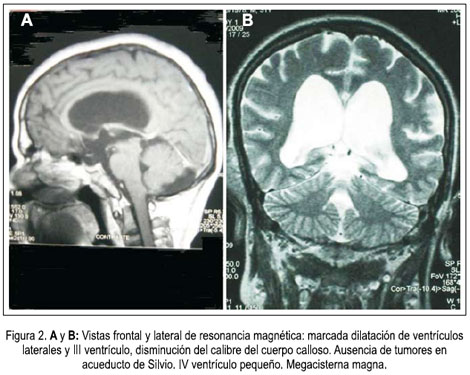
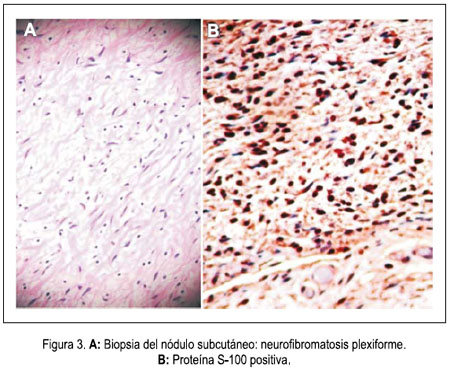
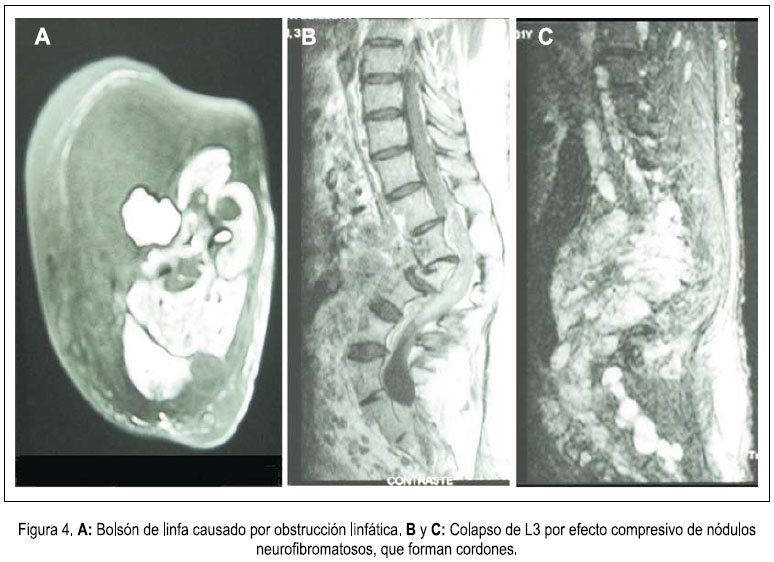
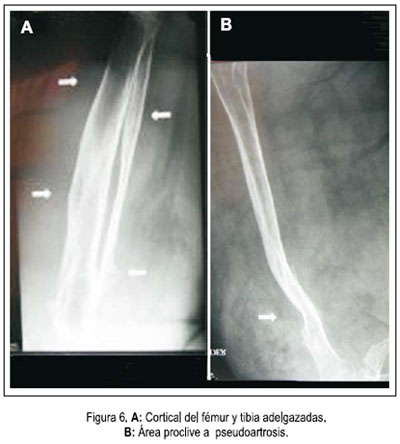
En la densitometría ósea se encontró osteoporosis generalizada de vértebras y huesos largos. La angiografía por tomografía espiral multicorte de miembros inferiores con contraste determinó elefantiasis plexiforme neurofibromatosa de miembro inferior derecho, múltiples neurofibromas en retro e intraperitoneo, displasia y osteoartrosis en fémur y tibia derecha. Dilataciones venosas de miembro inferior derecho, con retardo en la circulación. Fractura por aplastamiento en L3. Resonancia magnética de encéfalo demostró hidrocefalia triventricular moderada (laterales y tercer ventrículo), normotensa, no comunicante. Hipófisis comprimida contra el fondo selar. No se determinaba neoplasias. Biopsia de nódulo de muslo derecho: neurofibromatosis plexiforme. Proteína S-100 positivo. Biopsia de músculo oblicuo mayor y de conglomerado nodular retroperitoneal: neurofibromatosis plexiforme, proteína S-100 positiva. Survey óseo abdominopélvico: escoliosis dorsolumbar derecha. Deformidad de pelvis ósea, asimetría de excavación pelviana, protrusión acetabular con formación de neocótilo izquierdo. Numerosos quistes subperiósticos en regiones periacetabulares. Fémures invadidos por neurofibromas subperiósticos. Adelgazamiento de la cortical del fémur y tibia, cambios erosivos por neurofibromas. Tumoración intrabadominal multinodular. Elefantiasis en el miembro inferior derecho. Resonancia magnética de muslo, pierna y pié derecho: nódulos acordonados a lo largo del miembro inferior derecho, infiltrando músculos, más osteodistrofia, compatibles con NF1 plexiforme de miembro inferior derecho, y en menor grado del miembro inferior izquierdo. figuras 4, 5 y 6.
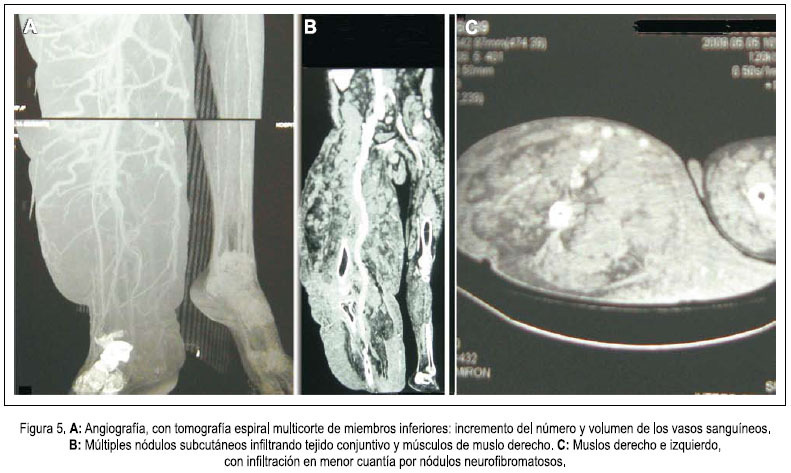
El paciente presentó signos típicos de NF1: a) más de 6 manchas café con leche mayores de 15 mm de diámetro; b) más de dos neurofibromas; c) pecas en axilas; d) más de dos nódulos de Lisch; e) adelgazamiento de la cortical de la tibia y fémur; f) trastornos cognitivos; g) familiar en primer grado con NF1 (1,2,5); elefantiasis neurofibromatosa, hidrocefalia y displasia ósea.
Los nódulos y tumoraciones nodulares en regiones intraabdominal, retroperitoneal y mediastinal en las resonancias magnéticas nucleares (RMN) se ajustaron a las descripciones de crecimiento intrafascicular de neurofibromas plexiformes a lo largo de los nervios espinales (6,7). Ocasionalmente, el crecimiento nodular es masivo, hipertrofiando difusamente los tejidos conectivos cercanos. La asimetría de los miembros inferiores, el sobrecrecimiento del miembro inferior derecho, así como de piel y vasos sanguíneos, hizo pensar inicialmente en la existencia de un síndrome Proteus (8), que fue descartado por la presencia de nódulos de Lisch, manchas café con leche y ausencia de nevus conectivo-cerebriformes, macrodactilia y clinodactilia. La asimetría de miembros es explicada por el descenso de la cadera derecha, en tanto el sobrecrecimiento del miembro inferior derecho es explicado por interacciones locales durante el desarrollo entre tejidos dérmicos y mesenquimatosos, por alteración primaria de la cresta neural, con ulterior hiperplasia o neoplasia de los mismos (9). La RMN de miembros inferiores mostró marcada infiltración nodular plexiforme de los músculos del muslo derecho y en menor cuantía del izquierdo. Según Holck (10), en la elefantiasis neurofibromatosa predominan los nódulos neurofibromatosos profundos (11,12), dando un aspecto voluminoso, paquidérmico, a su miembro afectado.
En lo que se refiere a otras complicaciones neurológicas de NF1, estas son variadas: cefalea, hidrocefalia, epilepsia, neurofibroma intraspinal, radiculopatías, afectación de las vainas de los nervios de las vías ópticas, meningiomas, tumor de las vainas de los nervios periféricos (11). La RMN cerebral mostraba marcada dilatación de los ventrículos laterales y del tercer ventrículo y en menor grado del cuarto ventrículo, no detectándose lesiones neoplásicas visibles. El cuerpo calloso y el séptum pellúcidum, presentes en toda su longitud, presentaban un aspecto laminar, a causa de compresión hidrocefálica. El manto cortical mostró algunos girus, de grosor por encima de lo normal. La hidrocefalia triventricular es frecuente en niños, asociándose mayormente a estenosis del acueducto de Silvio, con gliosis asociada (13,14). En estos casos, la hidrocefalia ocurre rápidamente en algunos pacientes, mientras que en otros evoluciona a lo largo de varios años, permaneciendo algunos asintomáticos, aunque sus fondos de ojo exhiban papiledema bilateral (15-17). Independiente de ello, estos pacientes han sido tratados exitosamente con shunts (17,18). Al obstruirse el acueducto, se dificulta la salida del líquido cefalorraquídeo, circula lentamente, sin inducir un cuadro brusco de hipertensión intracraneal (19,20-22). No obstante, por su procedencia, su labor como criador de cerdos y no beber agua clorada o hervida, es necesario descartar una neurocisticercosis racemosa (23).
Es probable que si la hidrocefalia del paciente ocurría a presión normal, la compresión cerebral explicaría los trastornos cognitivos. No obstante, se ha observado que la NF1 por sí misma se asocia a retardo mental y alteraciones del aprendizaje, por pérdida de la modulación de la actividad de la adenilciclasa, una proteína con un rol cardinal en la cognición (21,22).
Las imágenes de RMN mostraron gran número de nódulos plexiformes, infiltrando la región anterior de la columna vertebral lumbar, colapsando L3 y condicionando una marcada desviación de la región lumbosacra, con leve compresión de las raíces nerviosas de la cola de caballo. Las deformidades espinales severas pueden comprimir órganos torácicos internos, la médula espinal y promover talla corta, escoliosis, displasia del esfenoides y problemas dentales. De otro lado, las radiografías exhibieron adelgazamiento de la cortical de los huesos largos y remodelación de la cadera derecha, que lucía descendida. Al respecto, más de la mitad de pacientes con NF1 tiene manifestaciones esqueléticas primarias, no dependientes de neurofibromas adyacentes. Las últimas investigaciones asignan un rol al gen NF1 en el desarrollo y remodelación osteocartilaginoso, siendo que su deficiencia condiciona pseudoartrosis. La osteoporosis de huesos largos se explica por el uso de esteroides y reposo obligado. Con respecto a otros signos, es posible que el incremento de la frecuencia urinaria se deba a compresión tumoral de la vejiga. Aunque incompletamente estudiado, la TSH sugiere un hipotiroidismo subclínico. Los niveles bajos de cortisol correspondieron a una insuficiencia adrenal terciaria, a consecuencia de los esteroides recibidos desde hacía dos años (22). Finalmente, mencionaremos que la talla baja objetivada en este paciente guardaba relación con observaciones internacionales (23).
Se había planificado en este paciente realizar una desarticulación del miembro más afectado (24), insertar un shunt ventriculoperitoneal y extirpar las tumoraciones abdomino-pélvicas infiltrativas, además de solicitar western blot para cisticercosis, ventrículo-cisternografía, estudio neuropsicológico integral, administración de lovastatina para mejorar la cognición, pirfenidona (antifibrótico) y rapamicina, represor del crecimiento de células tumorales, procedimientos que no pudieron llevarse a cabo, ya que el paciente fue sometido a una desarticulación de todo el miembro inferior derecho, falleciendo 6 horas después en el postoperatorio, por coagulopatía de consumo, tras haber recibido más de 12 unidades de sangre en la intervención quirúrgica traumatológica.
AGRADECIMIENTOS
Al Dr. Jorge Yarinsueca G, Jefe de servicio de Medicina, Sala Santo Toribio, Hospital Nacional Dos de Mayo, Lima, Perú.
REFERENCIAS BIBLIOGRÁFICAS
1. Rosalie E Ferner. Neurofibromatosis 1. Eur J Hum Genet. Nature Publishing Group. 2007;15:131-8. [ Links ]
2. Huson SM. Neurofibromatosis: historical perspective, classification and diagnostic criteria. En: Huson SM, Hughes RAC, editors. The neurofibromatoses. A pathogenetic and clinical overview. London: Chapman and Hall. 1994a:1-22.
3. Campos P, Guillén D, Mera C, Botto C, Castañeda C. Trastornos neuroectodérmicos y epilepsia. Experiencia de 10 años en un Hospital General. Rev peru Neurol. 1999;5(1):6-10. [ Links ]
4. Dasgupta B, Dugan LL, Gutmann DH. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosc. 2003;23(26):8949-54. [ Links ]
5. Neurofibromatosis Conference Statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-8.
6. Huson SM. Neurofibromatosis 1: A clinical and genetic overview. En: Huson SM, Hughes RAC, editors. The neurofibromatoses. A pathogenetic and clinical overview. London: Chapman and Hall. 1994 b:160-203.
7. Fortman BJ, Kuszyk BS, Urban BA, Fishman EK. Neurofibromatosis type 1: A diagnostic mimicker at CT1. RadioGraphics. 2001;21:601-12. [ Links ]
8. Turner JT, Cohen Jr. MM, Biesecker LG. Reassessment of the Proteus syndrome literature: application of diagnostic criteria to published cases. Am J Med Genet. 2004;130:111-22. [ Links ]
9. Hughes RC. Neurological complications of neurofibromatosis. En: Huson SM, Hughes RAC, editors. The neurofibromatoses. A pathogenetic and clinical overview. London: Chapman and Hall. 1994:204-32. [ Links ]
10. Holck S, Medgyesi S, Darre E, Lassen M. Elephantiasis neuromatosa. Virchows Arch (Path Anat). 1984;404:427-34. [ Links ]
11. Creange A, Zeller J, Rostaing-Rigattieri S, Brugieres P, Degos J-D, Revuz J, Wolkenstein P. Neurological complications of neurofibromatosis type 1 in adulthood. Brain. 1999;122:473-81. [ Links ]
12. Stevens KJ, Ludman CN, Sully L, Preston BJ. Magnetic resonance imaging of elephantiasis neuromatosa. Skeletal Radiol. 1998;12:696-701. [ Links ]
13. Pascual-Castroviejo I, Pascual-Pascual SI, Velázquez-Fragua R, Viaño J, Carceller-Benito F. Aqueductal stenosis in the neurofibromatosis type1. Presentation of 19 infantile patients. Rev Neurol. 2007;45(1):18-21. [ Links ]
14. Brasfield RD, Das Gupta TK. Von Recklinghausen's disease: A clinic pathological study. Ann Surgery. 1972;175:86-104. [ Links ]
15. Balestrazzi P, de Gressi S, Donadio A, Lenzini S. Periaqueductal gliosis causing hydrocephalus in a patient with neurofibromatosis type 1. Neurofibromatosis. 1989;2(issue 5-6):322-5. [ Links ]
16. Pou-Serradell A, Ugarte-Elola AC. Hydrocepahlus in neurofibromatosis. Conribution of magnetic resonance imaging to its diagnosis, control and treatment. Neurofibromatosis. 1989;2(4):218-26. [ Links ]
17. Nieto T, Reyes P, Miller E. Neurocisticercosis espinal: a propósito de un caso. Rev méd hondur. 1994;62(4):164-6. [ Links ]
18. Riccardi VM, Eichner JE. Psychosocial aspects. En: Riccardi VM, Eichner JE, editors. Neurofibromatosis. Phenotype, natural history and pathogenesis. Baltimore: Johns Hopkins Univ Pr. 1986:150-68.
19. Weeber EJ, Sweatt JD. Molecular neurobiology of human cognition. Neuron. 2002;33(6):845-8. [ Links ]
20. Reed N, Gutmann DH. Tumorigenesis in neurofibromatosis: new insights and potential therapies. Trends Molec Med. 2001;7:157-62. [ Links ]
21. Huang L, Manning LDD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179-90. [ Links ]
22. Tóth M, Szúcs N, Rácz K,Varga I, Hüttl K, Perner F, Gláz E. Endocrinologic complications of neurofibromatosis type 1. Orv Hetil. 1996;137(31):1683-7. [ Links ]
23. Carmi D, Shohat M, Metzker A, Dickerman Z. Growth, puberty, and endocrine functions in patients with sporadic or familial neurofibromatosis type 1: a longitudinal study. Pediatrics. 1999;103(6 Pt 1):1257-62. [ Links ]
24. MacEwen GD. Orthopedic aspects of neurofibromatosis. En: Rubenstein AE, Korf BR, editors. Neurofibromatosis. A handbook for patients, families and health care professionals. New York: Thieme Medical Publ. 1990:25-141. [ Links ]
Manuscrito recibido el 22 de julio de 2009 y aceptado para publicación el 4 de setiembre de 2009.
Correspondencia:
Dr. Victor Mechán Méndez
Urbanización Palmeras de Villa Km 18,5
Panamericana Sur MzB Lote 1
Lima 9, Perú
Correo-e: victor.mechan@gmail.com

