










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkAnales de la Facultad de Medicina
Print version ISSN 1025-5583
An. Fac. med. vol.70 no.3 Lima Sept. 2009
Neurofibromatosis tipo 1 asociado a tumor maligno de la vaina de nervio periférico y a carcinoma de colon
Neurofibromatosis type 1 associated to peripheral nerve sheath and colon malignant tumor
Rubén Valle1, Víctor Valencia2,3, José Huamán-Muñante2,3
1 Médico Cirujano egresado de Facultad de Medicina, Universidad Nacional Mayor de San Marcos. Lima, Perú.
2 Profesor de la Facultad de Medicina, Universidad Nacional Mayor de San Marcos. Lima, Perú.
3 Hospital Nacional Arzobispo Loayza. Lima, Perú.
Resumen
La neurofibromatosis tipo 1 es una enfermedad autosómica dominante, producida por la mutación del gen de la neurofibrina, localizado en el cromosoma 17q11.2. Esta enfermedad presenta una diversidad de manifestaciones clínicas y predisposición al desarrollo de tumores, lo cual explica la elevada mortalidad. Presentamos el caso de una paciente con diagnóstico de neurofibromatosis tipo 1, quién desarrolló un tumor maligno de la vaina del nervio periférico y cáncer de colon. Se revisa la literatura y los factores clínicos, de imagen e histológicos que se asocian a la transformación maligna de los neurofibromas.
Palabras clave: Neurofibromatosis 1; neurofibroma; sarcoma; neoplasias; colon.
Abstract
Neurofibromatosis type 1 is a dominant autosomal disease caused by mutation of the neurofibrin gene located in chromosome 17q11.2. This disease presents a variety of clinical manifestations and predisposition to develop tumors, leading to high mortality. We report the case of a patient with neurofibromatosis type 1 diagnosis who developed a malignant tumor of peripheral nerve sheath and colon cancer. The authors review the literature as well as the clinical factors, imaging and histological features associated to neurofibromas malignant transformation.
Key words: Neurofibromatosis 1; neurofibroma; sarcoma; neoplasms; colon.
INTRODUCCIÓN
La neurofibromatosis tipo I (NF1), también llamada enfermedad de Von Recklinghausen, es una enfermedad autosómica dominante, de penetrancia completa y expresión fenotípica variable, que en 50% de los casos se produce por mutaciones esporádicas (1,2). El gen responsable de la enfermedad está localizado en el cromosoma 17q11.2 y, al estar mutado su producto, la neurofibrina, pierde la capacidad de supresor tumoral, originando la predisposición al desarrollo de diferentes neoplasias (3,4), por lo que estos pacientes presentan una incidencia de cáncer 3% mayor que la población general (2).
La incidencia es una en 2 500 a 3 300 personas (5), sin preferencia por el género o raza (1). Los criterios diagnósticos fueron establecidos por la National Institutes of Health Consensus Development Conference, en 1987, y para hacer el diagnóstico se requiere de la presencia de dos o más de los siete criterios clínicos siguientes: seis o más manchas café con leche que midan al menos 0,5 cm de diámetro antes de la pubertad o al menos 1,5 mm después de la pubertad, dos o más neurofibromas de algún tipo o un neurofibroma plexiforme, pecas en la región inguinal o axilar, gliomas en la vía óptica, dos o más nódulos de Lisch (hamartomas del iris), displasia del hueso esfenoides o displasia o adelgazamiento de la corteza de un hueso largo y un pariente de primer grado con NF1 (6).
Presentamos el caso de una paciente con el diagnóstico de NF1 que ingresó por presentar náuseas, vómitos, dolor a nivel de la articulación coxofemoral derecha y una tumoración en hipocondrio izquierdo. Además, durante su hospitalización presentó insuficiencia renal aguda (IRA) y un cuadro de hipertensión endocraneana (HTE).
CASO CLÍNICO
Paciente mujer de 30 años, natural y procedente de Ayacucho, acudió por manifestar náuseas y vómitos por 19 días y dolor en la articulación coxofemoral derecha, que comprometía la marcha desde hacía 4 semanas. Veinte días atrás se le realizó una colecistectomía laparoscópica, por dolor abdominal durante 5 meses en hemiabdomen superior, con predominio en el hipocondrio izquierdo. Como antecedente, señaló que había tenido convulsiones desde los 3 años y la última fue hacía un año y medio; la menarquía ocurrió a la edad de 19 años.
Al examen, era una mujer en regular estado general, de contextura delgada, con leve palidez de piel y con dificultad en la marcha. La frecuencia cardiaca era 86 por minuto, presión arterial (PA) 140/90, temperatura 36,8°C y frecuencia respiratoria 18 por minuto. En la piel, se observó manchas hipercrómicas e hipocrómicas (manchas café con leche) distribuidas en región anterior y posterior del tórax, lesiones nodulares de 1 cm de diámetro en tórax y región suprapalpebral derecha y pecas en región axilar e inguinal. El tejido celular subcutáneo estaba disminuido. En la evaluación cardiovascular, se auscultó un soplo holosistólico II/VI multifocal, acompañado de extrasístoles cardiacas. En el abdomen, se observaba cicatrices de la intervención quirúrgica y un tumor de 4 x 5 cm en el hipocondrio izquierdo, dependiente de partes blandas, móvil, bien delimitado y doloroso a la palpación superficial, el cual refiere que lo presentaba desde hacía un mes; sin embargo, el dolor en la región lo tenía desde hacía 5 meses. En el examen osteo-mio-articular mostraba dolor a la rotación externa e interna de la articulación coxofemoral derecha, asociado a leve disminución de la fuerza muscular, a predominio proximal; en el examen neurológico, la marcha se encontraba comprometida por el dolor y presentaba ampliación de la base de sustentación; así mismo, mostraba leve dismetría del dedo índice-nariz derecho. No se encontró alteraciones en el resto del examen.
La paciente cumplía con 5 de los 7 criterios clínicos de NF1, estando presentes: más de seis manchas café con leche que medían más de 1,5 mm de diámetro, más de dos neurofibromas, pecas en la región inguinal y axilar, más de dos nódulos de Lisch y un pariente de primer grado con NF1 (madre); sin embargo, nunca había sido diagnosticada de esta enfermedad. La madre de la paciente cumplía con tres criterios y refería que tenía familiares que presentaban las mismas manchas desde el nacimiento.
En los exámenes de laboratorio (tabla 1), la paciente ingresó con una azoemia que remitió aproximadamente a los 18 días de hospitalización. Además, presentó leucocituria, por lo que fue tratada con antibióticos, mostrando luego urocultivo de control negativo. Se realizó el descarte de feocromocitoma midiendo el nivel de metanefrinas en orina, por los signos clínicos presentes (PA=140/90, extrasístoles cardiacas, pérdida de peso) y la asociación existente entre NF1 y feocromocitoma, obteniendo un resultado normal. Por un evento de deposiciones con rasgos de sangre asociado a estreñimiento, se solicitó un estudio de sangre oculta, resultando negativo.
Por el cuadro de azoemia, se le realizó una ecografía renal, que informó riñones de dimensiones normales, con un pequeño cálculo en riñón derecho. La ecografía de partes blandas, en hipocondrio izquierdo mostraba tumoración sólida de 3,5 x 2,6 x 3,6 cm, de bordes bien definidos. La radiografía de columna lumbosacra informó esclerosis de platillos vertebrales L4 y L5, y la de pelvis, esclerosis de los techos acetabulares y de la articulación sacroiliaca derecha. En el examen oftalmológico, se evidenció los nódulos de Lisch (criterio clínico de NF1) y la ausencia de edema de papila. En la electromiografía del miembro inferior derecho no se observó signos patológicos.
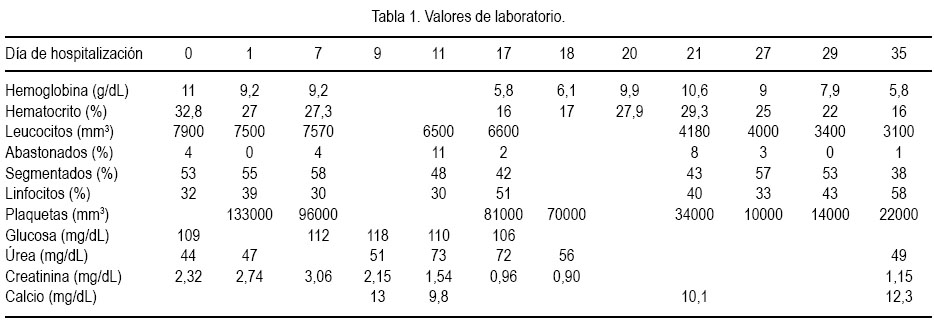
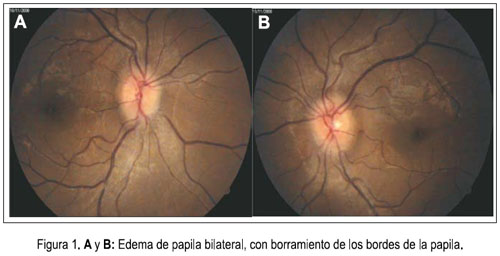
Al cuarto día de hospitalización, las náuseas y vómitos remitieron y empezó a tolerar mejor la vía oral. Sin embargo, el día 7 volvió a presentar náuseas y vómitos de contenido alimentario y al día siguiente se agregó cefalea intensa asociada a mareos, por lo que se repitió el examen oftalmológico y se constató edema de papila bilateral (figura 1). Se le tomó una tomografía axial computarizada (TAC) sin contraste, debido a la azoemia presente, y no se evidenció proceso expansivo u otra alteración cerebral estructural, siendo la paciente catalogada como un cuadro de hipertensión endocraneana benigna o pseudo tumor cerebral. Se inició tratamiento con manitol y dexametasona, con lo que el cuadro remitió. Posteriormente, la resonancia magnética nuclear mostró hamartomas cerebelosos y un angioma venoso subcortical en el lóbulo frontal izquierdo (figura 2).
En el noveno día de hospitalización, se solicitó calcio (Ca) para descartar un síndrome paraneoplásico y este resultó elevado, con valores normales de parathormona; sin embargo, el valor de Ca disminuyó en los análisis siguientes, al parecer asociado a los corticoides e hidratación con soluciones hipertónicas que recibía como tratamiento de la hipertensión endocraneana; al concluir este tratamiento, el Ca volvió a elevarse. La paciente ingresó con una bicitopenia (anemia y plaquetopenia) que, en el 19 día de hospitalización, concomitantemente a la remisión de la azoemia, se acentuó con una caída significativa de los valores de hemoglobina (5,8 mg/dL) y de plaquetas, por lo que se transfundió dos paquetes globulares. En el noveno día de hospitalización, durante la biopsia de la tumoración, hubo una pérdida de sangre de aproximadamente 400 mL. El conteo de reticulocitos evidenció un índice de producción de reticulocitos, que indicaba hipofunción medular, por lo que se le realizó aspirado de médula ósea y biopsia de hueso. Posteriormente, la paciente cursó con episodios de epistaxis espontánea en dos oportunidades, por lo que se le transfundió 4 unidades de plaquetas.
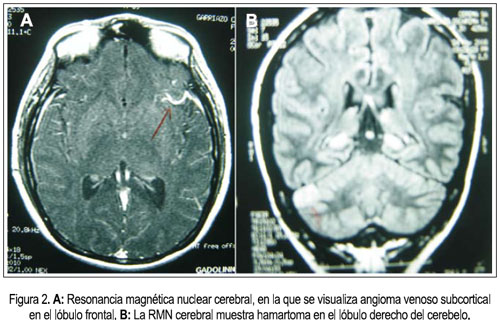
La biopsia de la tumoración del hipocondrio izquierdo señaló tumor de partes blandas, neoplasia maligna sarcomatoide con áreas de necrosis (figura 3: A y B). El aspirado de médula ósea informó médula ósea con presencia de células neoplásicas extramedulares, sugerente de mieloptisis (figura 3: C y D). Y la biopsia de hueso encontró tejido óseo infiltrado por neoplasia de células redondas-sarcomatoide; en la inmunohistoquímica, se la describió como neoplasia fusocelular compatible con características de tumor maligno de la vaina del nervio periférico, grado 1; panqueratina negativa; proteína S-100 con células ocasionales (+); ACL (-); calretinina con células ocasionales (+); KI-67 10 a 15%. Con estos resultados, la paciente fue transferida al Instituto Nacional de Enfermedades Neoplásicas (INEN), para tratamiento especializado.
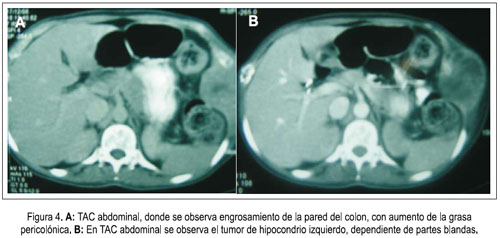
En el INEN, en una tomografía torácica realizada para ver la presencia de metástasis y estadiaje se observó múltiples lesiones osteolíticas en arcos costales y cuerpos vertebrales, lo que posteriormente produciría un colapso del séptimo cuerpo vertebral. La radiografía de la articulación coxofemoral derecha mostró lesiones líticas de 1 cm de diámetro, acompañadas de fractura patológica del cuello anatómico. En el día 38 de hospitalización en el INEN, se observó un patrón parenquimal inflamatorio en la radiografía de tórax, por lo que fue tratada como una paciente neutropénica febril, de foco pulmonar. En la TAC de abdomen se encontró engrosamiento del colon derecho y recto, así como, aumento de la grasa perirrectal (figura 4), por lo que se le realizó colonoscopia, que informó lesiones elevadas blanquecinas de aproximadamente de 5 mm que tapizaban toda la mucosa, con pequeñas áreas de mucosa normal (pancolitis); la biopsia señaló distorsión (dropout) de las criptas superficiales, con pérdida de epitelio, rodeado de infiltrado inflamatorio agudo moderado y necrosis estromal; algunas glándulas mostraban epitelio atípico y mitosis, algunas con morfología en anillo de sello. Se solicitó repetir el examen para obtener una mejor muestra. En biopsia de pared abdominal diagnosticaron sarcoma fusocelular de grado alto. La panqueratina fue positiva.
En el día 54 de hospitalización en el INEN, la paciente cursó con dolor, distensión abdominal y fiebre (foco abdominal), y la radiografía de abdomen fue informada con distensión de asas intestinales, asociada a niveles hidroaéreos. Al día siguiente, la distensión abdominal aumentó, con mayor frecuencia respiratoria y tendencia a hipotensión. Revisada la lámina de colon por otro patólogo, volvió a informar carcinoma en células en anillo de sello. Luego, la radiografía de tórax fue informada con opacidad homogénea que ocupaba la base del hemitórax derecho, que ascendía parcialmente por la pared lateral hasta el ápice. Posteriormente, la distensión abdominal aumentó y la hipotensión se volvió refractaria a los vasopresores. Finalmente, la paciente falleció, consignándose como causa básica de muerte al cáncer de colon de células de anillo de sello, con infiltrado de médula ósea y metástasis ósea.
DISCUSIÓN
La NF1 es un desorden autosómico dominante, que se transmite en 50% de los casos de forma hereditaria; presenta penetrancia completa. Las manifestaciones clínicas varían con la edad y la expresión fenotípica puede ser diferente en un mismo grupo familiar (1,2,5). En el caso presentado, la paciente tenía familiares ascendentes y descendentes que tenían las mismas lesiones en la piel; sin embargo, nunca habían sido diagnosticados de NF1 y al parecer nunca presentaron una comorbilidad considerable.
Al ingreso, la paciente mostró insuficiencia respiratoria aguda (IRA) y posteriormente un cuadro de hipertensión endocraneana; al inicio, solo se realizó TAC sin contraste, por la azoemia presente, sin observar alguna alteración especial. Posteriormente, se indicó RMN cerebral, en la que se observó hamartomas cerebelosos y un angioma venoso, lesiones que han sido descritas en estos pacientes (2,7). No se pudo determinar la causa de la IRA. Sin embargo, este cuadro puede haber predispuesto al desarrollo de la hipertensión endocraneana (8), no pudiendo considerarse una hipertensión endocraneana idiopática, por la presencia de las lesiones vasculares en la RMN (8,9). El tratamiento de la hipertensión endocraneana (corticoides e hidratación con sales hipertónicas), provocaron la disminución de la hipercalcemia, lo que sugería desde un principio la presencia de una neoplasia.
La NF1 se encuentra dentro del síndrome de predisposición tumoral (10) y, entre las neoplasias más frecuentes a las que predispone, se hallan los tumores de la vaina de los nervios periféricos, feocromocitoma, leucemia mieloide, astrocitomas pilocíticos, sarcomas, tumores espinales, rabdomiosarcomas (2,11-13), y tumores del estroma intestinal, los cuales son los tumores mesenquimales más frecuentes del tracto gastrointestinal (14,15). Entre los tumores menos frecuentes están los de mama, pulmón, riñón y de colon, siendo su frecuencia incluso menor que en la población normal (16).
Los tumores malignos de la vaina del nervio periférico (TMVNP) representan la neoplasia más frecuentemente asociada a esta enfermedad y se pueden desarrollar de neurofibromas preexistentes o de novo (11), incluso se ha reportado el crecimiento de un TMVNP, sobre la cicatriz operatoria en un paciente con diagnóstico de NF1 (17). Existe una fuerte asociación entre NF1 y los TMVNP (12,18); el riesgo de desarrollar un TMVNP en un paciente con NF1 es de 8 a 13% (18,19), riesgo muy elevado en comparación del que existe en la población general. Si bien muchos de los neurofibromas llegan a malignizarse entre los 20 y 50 años (20), en muchas ocasiones estas neoplasias se desarrollan a partir de la infancia. En un estudio, la prevalencia de NF1 en niños con TMVNP fue de 43% (12). Si bien estos tumores pueden estar presentes sin causar sintomatología clínica (13), en otros casos producen escoliosis, neurofibromas localizados y neurofibromas de tejidos superficiales, por lo que se ha propuesto que el tamizaje con RMN a toda la columna debería ser aplicada en pacientes con NF1 desde edades tempranas (21).
A raíz de la asociación reconocida entre la NF1 y los TMVNP, se ha planteado diferentes formas de reconocer -a través de la clínica, imágenes e histología- cuando un neurofibroma se ha malignizado. Por ejemplo, se ha reconocido que los síntomas como el dolor, crecimiento tumoral y los signos neurológicos se encuentran asociados con la transformación maligna del tumor (22,23). Entre algunos signos en la resonancia encontramos: tamaño mayor de 5 cm, bordes mal definidos, carecer de hipointensidad central en imágenes T2 y heterogenicidad con necrosis central (24-26). Valeyrie, en su trabajo, definió como displasia del neurofibroma a la presencia de alta celularidad y células atípicas y encontró que la displasia se asociaba a los síntomas neurológicos. Concluyó que la displasia podría ser un precursor premaligno antes de la transformación en TMVNP (27). Ramanathan observó que un tamaño tumoral mayor de 10 cm y un grado histológico alto son factores pronósticos que influyen en la supervivencia global en los pacientes con NF1 (11).
En el presente caso, la paciente tuvo un crecimiento tumoral de 6,5 cm en 2 semanas (control por ecografía) -que pudo haber aumentado por el componente inflamatorio producido después de la biopsia-, un cuadro neurológico de hipertensión endocraneana, así como dolor en la región del tumor, aproximadamente 4 meses antes del crecimiento tumoral, lo que confirma que los signos clínicos anteriormente mencionados se asociaban a malignidad tumoral. De la misma manera, el tumor era de grado de diferenciación celular alto, lo cual devino en la evolución fatal de la enfermedad.
La paciente, aparte de tener un TMVNP confirmado por histología e inmunohistoquímica, también presentó un cáncer de colon en células de anillos de sello. Sin embargo, la primera lectura no fue del todo concluyente, por lo que se solicitó una nueva muestra; por la evolución de la enfermedad, se hizo imposible una nueva toma, realizándose una segunda lectura por otro patólogo, quien refrendó el diagnóstico. Las neoplasias del tracto gastrointestinal son poco frecuentes en los pacientes con NF1 e incluso llegan a ser menos que en la población general (16); las más frecuentes son los tumores del estroma intestinal (14,15). En la literatura se ha comunicado 4 casos de adenocarcinomas de colon asociados a NF1 (28) y se ha encontrado 7 casos asociados a adenocarcinomas de intestino delgado; incluso, se ha llegado a describir dos adenocarcinomas separados en la región ileal, en un mismo paciente (29,30). La presencia de dos tumores de distinto origen no ha sido informada y esto nos muestra la alta susceptibilidad que confiere esta enfermedad para el desarrollo de neoplasias.
Entre las características de la inmunohistoquímica de la biopsia de médula ósea, vemos que era panqueratina negativa, lo cual nos orienta a un tumor que no era de origen epitelial, y concuerda con su naturaleza de TMVNP. Sin embargo, en la biopsia de la pared abdominal, el resultado de la misma muestra coincide con la naturaleza del tumor, pero menciona que es panqueratina positiva, lo cual podría ser un resultado falso positivo. El KI-67, que indica el nivel de proliferación celular del tumor, tuvo un valor de 10 a 15%, es decir, bajo a comparación del crecimiento tan acelerado que tenía el tumor.
En el Perú, no se conoce exactamente la prevalencia de esta entidad, ni en qué regiones se presentan los casos de transmisión genética. La paciente era natural y procedente de Ayacucho y, por las descripciones hechas, su bisabuelo tenía la enfermedad. Esta llegó a manifestarse hasta la cuarta generación descendente, sin saber ningún miembro de la familia que tenía o era portador de la enfermedad, ya que al parecer ninguno había presentado algún tipo de complicación. En este caso de NF1 de transmisión hereditaria, la construcción de un árbol genealógico hubiese sido de mucha importancia; sin embargo, la disgregación familiar que existía en la familia de la paciente hizo difícil construir un árbol genealógico confiable.
En el presente caso, se observa la variabilidad de manifestaciones clínicas que pueden desarrollar los pacientes con NF1, lo que obliga a un manejo multidisciplinario entre las especialidades, donde la consejería genética constituye una parte fundamental. Así mismo, se pone de manifiesto lo poco estudiada esta entidad en el Perú, ya que no se encontró bibliografía científica nacional que tuviera como tema de estudio a la enfermedad de Von Recklinghausen.
Sugerimos que, por el carácter heredo familiar de esta entidad, el abordaje debe ser desarrollado desde edades tempranas entre pacientes y familiares, con una pesquisa exhaustiva de los signos, prestando atención a los manifestaciones clínicas que van apareciendo conforme avanza la edad, haciendo uso de herramientas como la RMN, que permite conocer la presencia de neurofibromas asintomáticos. Posteriormente, en la edad adulta (20 a 40 años) se debería abordar el control de los neurofibromas, por el riesgo de malignización que existe en este grupo etáreo y que empobrece el pronóstico (31). Finalmente, se debe reconocer a los signos de dolor, crecimiento tumoral y signos neurológicos como predictores de malignidad, para una toma de decisiones más oportuna.
REFERENCIAS BIBLIOGRÁFICAS
1. Ward BA, Gutmann DH. Neurofibromatosis 1: From lab bench to clinic. Pediatr Neurol. 2005;32:221-8. [ Links ]
2. Tonsgard JH. Clinical manifestations and management of neurofibromatosis type 1. Semin in Pediatr Neurol. 2006;13:2-7. [ Links ]
3. Cawthon RM, Weiss R, Xu G, Viskochil D, Culver M, Stevens J, et al. A major segment of neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62(1):193-201. [ Links ]
4. Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh HM, Saulino AM, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181-6. [ Links ]
5. Friedman JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999;89:1-6. [ Links ]
6. National Institutes of Health Consensus Development Conference. Neurofibromatosis: conference statement. Arch Neurol. 1988;45:575-8. [ Links ]
7. Fletcher CD, Mckee PH. Soft tissue tumours. En: McGee JO, Isaacson Pg, Wright NA. Oxford Textbook of Pathology. Oxford: Oxford University Press, 1992; p. 2124. [ Links ]
8. Friedman D, Jacobson D. Diagnostic criteria for idiopathic intracranial hypertension. Neurology. 2002;59(10):1492-5. [ Links ]
9. Randhawa S, Van Stavern GP. Idiopatic intracranial hypertension (pseudotumor cerebri). Curr Op Ophthalmol. 2008;19:445-53. [ Links ]
10. Ferner RE. Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspective. Lancet Neurol. 2007;6(4):340-51. [ Links ]
11. Ramanathan R, Thomas M. Malignant peripheral nerve sheath tumours associated with von Recklinghausens neurofibromatosis. Eur J Surg Oncol. 1999;25:190-3. [ Links ]
12. Ferrari A, Bisogno G, Casanova A, DAngelo P, Pierani P, Zanetti I. Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer. 2007;109:1406-12. [ Links ]
13. Khong PL, Goh WH, Wong VC, Fung CW, Ooi GC. MR imaging of spinal tumors in children with neurofibromatosis I. Am J Roentgenol. 2003;180(2):413-7. [ Links ]
14. Takazawa Y, Shakurai S, Sakuma Y, Ikeda T, Yamaguchi J, Hashizume Y, et al. Gastrointestinal stromal tumors of neurofibromatosis type I (Von Recklinghausens Disease). Am J Surg Pathol. 2005;29(6):755-63. [ Links ]
15. Maertens O, Prenen H, Debiec-Rychter M, Wozniak A, Sciot R, Pauwels P, et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet. 2006;15(6):1015-23. [ Links ]
16. Sonrensen SA, Mulvihill JJ, Nielsen A. Long term follow-up of von Recklinghausen neurofibromatosis: survival and malignant neoplasms. N Engl J Med. 1986;314:1010-5. [ Links ]
17. García B, Ferrer M, Geylor A. Tumor maligno de la vaina de nervio periférico y neurofibromatosis. Rev Venez Oncol. 2008;20(1):34-7. [ Links ]
18. Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311-4. [ Links ]
19. Ferner RE, Gutman DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62:1573-7. [ Links ]
20. Woodruff JM. Pathology of tumors of the peripheral nerve sheath in type 1 neurofibromatosis. Am J Med Genet. 1999;89:23-30. [ Links ]
21. Egelhoff JC, Bates DJ, Ross JS, Rothner AD, Cohen BH. Spinal MR findings in neurofibromatosis type 1 and 2. AJNR. 1992;3:1071-7. [ Links ]
22. Valeyrie-Allanore L, Ismaïli N, Bastuji-Garin S, Zeller J, Wechsler J, Revuz J, Wolkenstein P. Symptoms associated with malignancy of peripheral nerve sheath tumours: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153:79-82. [ Links ]
23. King AA, Debaun MR, Riccardi VM, Gutmann DH. Malignant peripheral nerve sheath tumors in neurofibromatosis 1. Am J Med Genet. 2000;93:388-92. [ Links ]
24. Lin J, Martel W. Cross-sectional imaging of peripheral nerve sheath tumors: characteristic signs on CT, MR imaging, and sonography. AJR. 2001;176:75-82. [ Links ]
25. Bhargava R, Parham DM, Lasater OE, Chari RS,Chen G, Fletcher BD. MR imaging differentiation of benign and malignant peripheral nerve sheath tumors: use of the target sign. Pediatr Radiol. 1997;27:124-9. [ Links ]
26. Levine E, Huntrakoon M, Wetzel LH. Malignant nerve-sheath neoplasms in neurofibromatosis: distinction from benign tumors by using imaging techniques. AJR. 1987;149:1059-64. [ Links ]
27. Valeyrie-Allamore L, Ortonne N, Lantieri L, Ferkal S, Wechsler J, Bagot M, et al. Histopathologically dysplastic neurofibromas in neurofibromatosi 1: diagnostic criteria, prevalence and clinical significance. Br J Dermatol. 2008;158(5):1008-12. [ Links ]
28. Wood JJ, Longman RJ, Rooney N, Loveday EJ, Roe AM. Colonic vascular anomalies and colon cancer in neurofibromatosis: report of a case. Dis Colon Rectum. 2008;51(3):360-2. [ Links ]
29. Joo YE, Kim HS, Choi SK, Rew JS, Park CS, Kim SJ. Primary duodenal adenocarcinoma associated with neurofibromatosis. J Gastroenterol. 2002;37:215-9. [ Links ]
30. Hwangbo S, Kim J, Kim H, Kang C, Lee H. Two separated ileal adenocarcinomas in neurofibromatosis type 1. Yonsei Med J. 2007;48(6):1039-42. [ Links ]
31. Merchán R, Cacabelos P, Delgado C, Alañá M. Neurofibromatosis con metástasis pulmonares en paciente con neurofibromatosis tipo1. An Med Int. 2008;25(3):152. [ Links ]
Manuscrito recibido el 2 de junio de 2009 y aceptado para publicación el 18 de agosto de 2009.
Correspondencia:
Dr. Rubén Eliseo Valle Rivadeneyra
Jr. Filadelfia 2365, San Martín de Porres
Lima 31, Perú
Correo-e: ruben_vr12@hotmail.com

