










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. v.71 n.1 Lima ene./mar. 2010
Síndrome de costillas cortas y polidactilia: displasia esquelética fetal incompatible con la vida
Short rib polydactyly syndrome: lethal skeletal dysplasia
Erasmo Huertas1a,2, Jaime Íngar1a,3, Guiselle Gutiérrez1b,2, Eva María Quiñones3
1 Instituto Nacional Materno Perinatal. Lima, Perú.
2 Facultad de Medicina, Universidad Nacional Mayor de San Marcos. Lima, Perú.
3 Facultad de Medicina, Universidad Particular San Martín de Porres. Lima, Perú.
a Unidad de Medicina Fetal, b Servicio de Patología
Resumen
El síndrome costillas cortas polidactilia es una categoría descriptiva para un grupo de displasias esqueléticas incompatibles con la vida, caracterizadas por tórax estrecho, costillas extremadamente pequeñas, micromelia, polidactilia y anomalías viscerales. Las 4 variantes establecidas son SRPS I (tipo Saldino-Noonan), SRPS II (tipo Majewski), SRPS III (tipo Verma-Naumoff) y SRPS IV (tipo Beemer-Langer). Se piensa que todas las variantes son heredadas en forma autosómica recesiva. Debido a la frecuente superposición fenotípica, existe controversia si las variantes son debidas a expresión variable o a heterogeneidad genética. Presentamos un caso de un feto masculino, con signos ecográficos característicos de displasia esquelética, tales como micromelia, tórax estrecho en forma de campana, hipertelorismo, implantación baja de las orejas y polidactilia, que nació de parto vaginal y falleció a los 2 días de vida, producto de insuficiencia respiratoria, la cual presentó desde el nacimiento.
Palabras clave: Síndrome de Costilla pequeña y polidactilia; displasias óseas; ultrasonografía.
Abstract
Short rib-polydactyly syndrome is a descriptive category for a group of lethal skeletal dysplasias characterized by hypoplastic thorax, short ribs, short limbs, polydactyly, and visceral abnormalities. The 4 established variants are SRPS I (Saldino-Noonan type), SRPS II (Majewski type; 263520), SRPS III (Verma-Naumoff type; 263510), and SRPS IV (Beemer-Langer type; 269860). All variants are thought to be inherited in autosomal recessive pattern. Because of the frequent phenotype overlap there is controversy as to whether the variants are due to variable expression or to genetic heterogeneity. We present a case of a male fetus, with typical ultrasound markers of skeletal dysplasia such as micromelia, bell shaped narrow thorax, hypertelorism, low implantation ears and polidactily who was born by vaginal delivery and died two days later due to respiratory insufficiency present since birth.
Key words: Short rib-polydactyly syndrome; bone diseases, developmental; ultrasonography.
INTRODUCCIÓN
Las displasias esqueléticas constituyen un grupo heterogéneo de enfermedades del hueso, tanto del crecimiento como de su composición. Hasta la fecha, hay 372 subtipos ubicados en 37 grupos, según la Nosología y Clasificación de los Desórdenes Esqueléticos Genéticos (1). Aunque muchas displasias son letales, algunos individuos afectados sobreviven y llevan una vida productiva sin la intervención de terapia. La prevalencia al nacimiento de las displasias esqueléticas, excluyendo las amputaciones de miembros y reconocibles en el periodo neonatal, ha sido estimada en 2,4/10 000 nacimientos (2). Existe el diagnóstico molecular para muchas de estas patologías. Sin embargo, la valoración ecográfica prenatal sigue siendo el método de excelencia para la aproximación diagnóstica y seguimiento de las displasias óseas. Solo algunas displasias pueden ser detectadas con precisión en la ecografía prenatal. Unas cuantas comparten una característica básica, como el acortamiento de los huesos largos, lo cual presenta un desafío para el ecografista, diferenciar entre restricción del crecimiento intrauterino o un tipo de enanismo; pero, lo más importante es saber diferenciar si esta patología es letal o no, para lo cual se requiere un estudio secuencial de toda la anatomía fetal. Si bien la ecografía permite medir los huesos largos desde la décima semana de gestación, y las deformidades pueden ser detectadas desde el segundo trimestre, un diagnóstico tardío de una condición letal aumenta innecesariamente los riesgos para la salud física y mental de la paciente. Finalmente, el diagnóstico definitivo es patrimonio de una evaluación completa posnatal, que incluye el examen genético clínico, estudio radiográfico y estudio anatomopatológico. En vista de la extremadamente baja frecuencia de esta condición, describimos el primer caso de síndrome de costillas cortas polidactilia registrado en el Instituto Nacional Materno Perinatal (INMP) de Lima.
CASO CLÍNICO
Paciente de 18 años, natural de Cajamarca, procedente del distrito del Rímac, secundigesta nulípara, sin antecedentes médico-quirúrgicos de importancia, quien acudió al Instituto Nacional Materno Perinatal (INMP), el 3 de junio de 2009, referida del centro de salud donde se atendía, con el diagnóstico de asimetría fetal d/c malformaciones fetales, a las 37 semanas de gestación, según fecha de última regla (FUR).
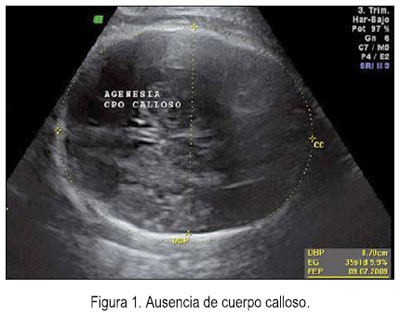
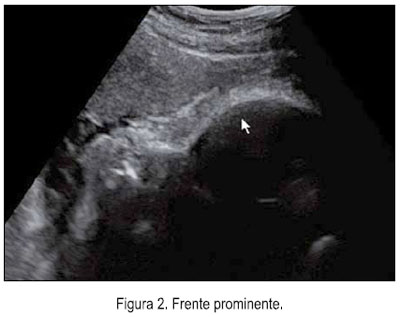
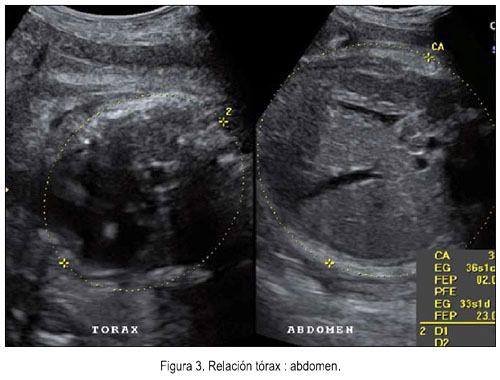
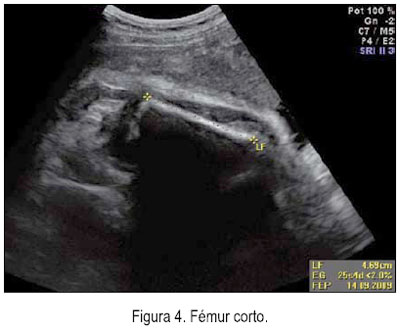
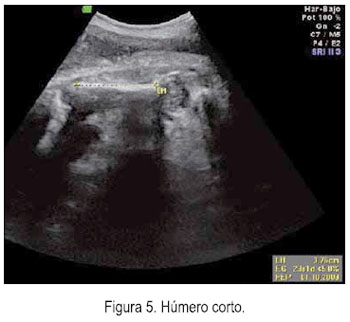
El 5 de junio se le realizó una ecografía en la unidad de medicina fetal de INMP, encontrándose un feto masculino con ausencia del cuerpo calloso (figura 1), frente prominente (figura 2), tórax estrecho (relación tórax/abdomen de 0,3) (figura 3) y marcado acortamiento de los huesos largos (figuras 4 y 5). Longitud de fémur 47 mm (consistente con 25 semanas) y longitud de húmero 38 mm (consistente con 23 semanas). Se concluyó como gestación única activa de 36 semanas por biometría de cabeza y abdomen, más displasia esquelética, probable displasia tanatofórica, con mal pronóstico perinatal.
El 9 de junio acudió a su segundo control prenatal (CPN), donde se explicó los hallazgos a la paciente y se indicó nuevo control en una semana. El 16 de junio fue vista en consultorios externos, esta vez con una intercurrencia, traqueítis aguda, por lo que se le indicó ambroxol y nuevo CPN en 7 días. El 23 del mismo mes, se observó persistencia de la tos, por lo que se realizó interconsulta a medicina interna, para descartar tuberculosis pulmonar.
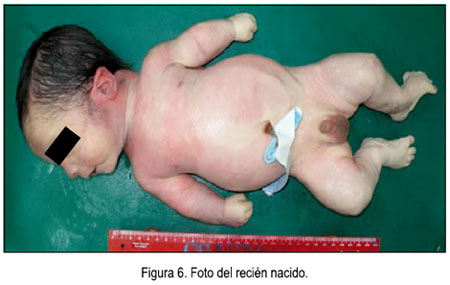
El 25 de junio la paciente ingresó por emergencia refiriendo contracciones uterinas espontáneas y 3 cm de dilatación al tacto vaginal; evolucionó favorablemente y tuvo parto vaginal de feto vivo de sexo masculino, con peso de 3145 g, talla 46 cm, perímetro cefálico 34 cm, perímetro torácico 32,5 cm, Ápgar 6 al minuto y 8 a los 5 minutos.
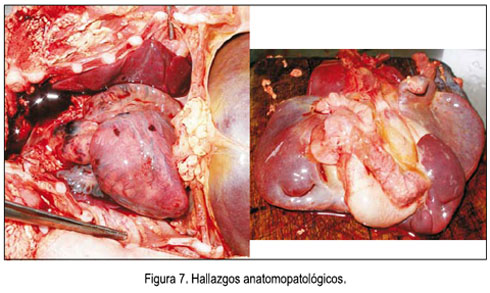
El examen físico del recién nacido reveló implantación baja de la oreja, criptorquidia unilateral derecha, polidactilia en miembros superiores e inferiores, extremidades cortas superiores e inferiores y cianosis periférica. Por presentar síndrome de distrés respiratorio moderado, fue hospitalizado en la unidad de cuidados intensivos neonatales, donde falleció a los 2 días de vida, por dificultad respiratoria. Se realizó el respectivo estudio posmórtem, que concluyó: recién nacido masculino de 32 semanas de gestación posfecundación, según antropometría, que corresponde a 34 semanas por FUR, de tamaño mayor al percentil 90 para peso y para perímetro cefálico, fallecido por posible sepsis u otra causa que se determinaría ulteriormente (figura 6). No se envió la placenta para estudio. Se sugirió control por consultorio externo de genética, para diagnóstico y consejería a los padres. Los hallazgos anatomopatológicos incluían situs inverso (figura 7), poliesplenia, ascitis, hipertelorismo, cuello corto, tórax estrecho, micropene de 2,5 cm, testículos descendidos y polidactilia.
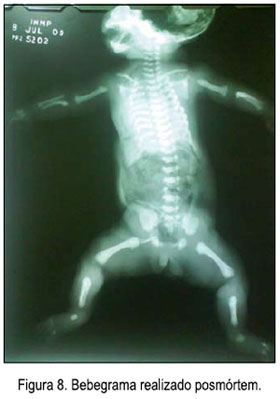
El bebegrama realizado posmórtem reveló micromelia y costillas cortas (figura 8). El cariotipo neonatal realizado mostró un varón normal (46 XY).
El síndrome costillas cortas polidactilia (SCCP) es una categoría descriptiva para un grupo de displasias esqueléticas letales caracterizado por tórax estrecho, costillas extremadamente pequeñas, micromelia, polidactilia y anomalías viscerales. La base de datos electrónica OMIM® -online mendelian inheritance in man (http://www.ncbi.nlm.nih.gov/omim)- la subdivide en 4 variantes, existiendo hasta la fecha controversia acerca si son entidades diferentes o partes de un espectro continuo con expresividad variable. En la tabla se puede apreciar las características distintivas de cada tipo.
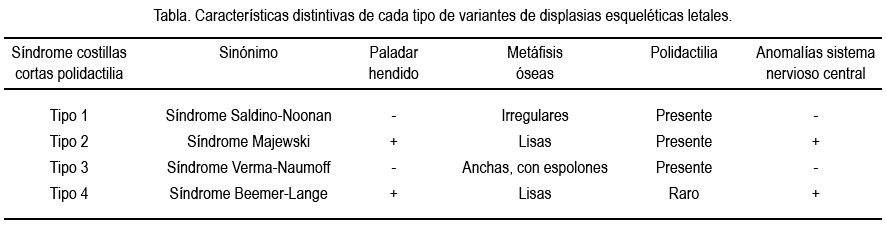
No se ha podido precisar la incidencia de este síndrome, en razón de su rareza, existiendo en la literatura solo reportes de casos o de series de casos (3). La prevalencia global de displasias esqueléticas al nacer es de 2,4 x 10 000 (2,4). Tampoco se ha observado predominio de un sexo sobre otro, pero sí ambigüedad sexual en una proporción importante de casos (5,6). El caso que motiva el presente reporte fue de un feto de sexo masculino, pero con micropene de 2,5 cm y testículos descendidos.
Si bien es cierto que el diagnóstico de SCCP fue hecho luego del nacimiento, en virtud a los hallazgos radiológicos y anatomopatológicos, es de destacar que el diagnóstico de displasia esquelética letal se realizó prenatalmente en base a la relación circunferencia torácica /circunferencia abdominal, la misma que se encontraba por debajo de 0,6, que es considerada un criterio de letalidad, ya que predice con bastante precisión hipoplasia pulmonar letal, independiente de la edad gestacional (7). Es necesario destacar que el hallazgo ecográfico de una displasia esquelética durante la evaluación de rutina de una gestación de tercer trimestre no debe distraernos de realizar un estudio sistemático y detallado de toda la anatomía fetal, de modo que no pasen inadvertidas alteraciones como situs inverso, poliesplenia, ascitis y polidactilia, presentes en el caso en discusión.
Es importante la consejería genética a los padres del bebe afectado, ya que el riesgo de recurrencia de esta patología es de 25%, debido a que tiene un patrón de herencia autosómica recesiva (8).
Otro aspecto crucial es que todos los recién nacidos con diagnóstico de displasia esquelética tengan evaluaciones posmórtem adecuadas (estudio radiológico, autopsia, estudio citogenético) que respalden científicamente la subsecuente consejería.
El diagnóstico final del caso presentado sería de un SCCP tipo 1 o Saldino-Noonan, en razón que presentó, además de la micromelia, tórax estrecho, anomalías cardiacas (situs inverso) y viscerales (poliesplenia), ausencia de paladar hendido, metáfisis óseas irregulares, polidactilia y ausencia de anomalías del sistema nervioso central (9,10).
Los primeros reportes de este tipo de displasia esquelética datan de 1972 (11), aunque ya un año antes Majewsky había comunicado el primer caso de SCCP (12). El manejo de esta patología, en países como el nuestro, donde el aborto eugenésico no es legal, consiste en atender el parto vaginal y no intentar maniobras de resucitación ni intubación del neonato, ya que el deceso se producirá inexorablemente en pocas horas, debido a la hipoplasia pulmonar severa.
REFERENCIAS BIBLIOGRÁFICAS
1. Superti-Furga A, Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am J Med Genet A. 2007;143:1-18. [ Links ]
2. Camera G, Mastroiacovo P. Birth prevalence of skeletal dysplasias in the Italian multicentric monitoring system for birth defects. En: Papadatos CJ, Bartsocas CS (eds). Skeletal Dysplasias. New York: Alan R Liss; 1982. p. 441-9. [ Links ]
3. Silva S, Jeanty P. Short rib polydactyly syndromes [Internet]. Nashville, Tennessee: TheFetus.net; c1990-2010 [citado el 4 de enero de 2010]. Disponible en: http://www.thefetus.net/page.php?id=372
4. Stoll C, Dolt B, Roth MP, Alembick Y. Birth prevalence rates of skeletal dysplasia. Clin Genet. 1989;35(2):88-92. [ Links ]
5. Yang SS, Lin CS, Al Saadi A, Nangia BS, Bernstein J. Short rib-polydactyly syndrome, type 3 with chondrocytic inclusions: report of a case and review of the literature. Am J Med Genet. 1980;7:205-13. [ Links ]
6. Bernstein R, Isdale J, Pinto M, Du Toit Zaaijman J, Jenkins T. Short rib-polydactyly syndrome: a single or heterogeneous entity? A re-evaluation prompted by four new cases. J Med Genet. 1985;22:46-53[ [ Links ]STANDARDIZEDENDPARAG]
7. Yoshimura S, Masuzaki H, Gotoh H, Fukuda H, Ishimaru T. Ultrasonographic prediction of lethal pulmonary hypoplasia: comparison of eight different ultrasonographic parameters. Am J Obstet Gynecol. 1996;175:477-83. [ Links ]
8. Meng HW, Pao LK, Shio JL. Prenatal diagnosis of recurrence of short rib polydactyly syndrome. Am J Med Genet. 1995;55:279-84. [ Links ]
9. Verma A. Short rib polydactyly syndrome type I (Saldino-Noonan syndrome). Indian Pediatr. 2005;42:389 [ Links ]
10. Lowry R, Wignall N. Saldino-Noonan short rib polydactyly dwarfism syndrome. Pediatrics. 1975;56:121-3. [ Links ]
11. Saldino RM, Noonan CD. Severe thoracic dystrophy with striking micromelia, abnormal osseous development, including the spine, and multiple visceral anomalies. Am J Roentgenol. 1972;114:257-63. [ Links ]
12. Majewski F, Pfeiffer RA, Lenz W, Muller R, Feil G, Seiler R. Polydactyly, short limbs, and genital malformations - a new syndrome? Z Kinderheilkd. 1971;111:118-38. [ Links ]
Manuscrito recibido el 4 de enero de 2010 y aceptado para publicación el 23 de febrero de 2010.
Correspondencia:
Dr. Erasmo Huertas Tacchino
Calle Carlos Pane 143
Lima 5, Perú.
Correo-e: erasmohuertas@hotmail.com

