










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. v.71 n.4 Lima dic. 2010
Hipertensión arterial - Aspectos genéticos
Arterial hypertension - Genetics
María Isabel Quiroga de Michelena1
1 Profesor Principal, Facultad de Medicina Alberto Hurtado, Universidad Peruana Cayetano Heredia.
Resumen
La regulación de la presión arterial (PA) es un proceso complejo, con intervención de múltiples variantes genéticas y epigenéticas que son objeto de la presente revisión. La disrupción del equilibrio entre los factores mencionados y su interacción con el ambiente lleva a la hipertensión arterial (HTA), que en la gran mayoría de las veces es primaria o esencial. Se revisa los genes involucrados, los raros cuadros de HTA de origen monogénico, y las perspectivas en farmacogenómica, así como las tendencias hacia la medicina personalizada y con mayor participación activa del paciente en el cuidado de su propia salud.
Palabras clave: Hipertensión, genética; genes; presión sanguínea; genética médica; genética de población.
Abstract
Regulation of blood pressure depends on complex mechanisms that include multiple genetic and epigenetic variations. There is a delicate balance between genetic, epigenetic and environmental factors that, when disrupted, lead to hypertension. In most cases blood hypertension is primary or essential, and rarely is inherited through mutation of a single gene. This review will focus on genes involved in multifactorial blood hypertension and in the rare cases of monogenic hypertension. The pharmacogenomic advances and perspectives in the era of genomic medicine will also be reviewed.
Keywords: Hypertension, genetics; genes; blood pressure; genetics medical; genetics population.
INTRODUCCIÓN
La regulación de la presión arterial (PA) es un proceso fisiológico complejo, en el que intervienen factores genéticos, epigenéticos y ambientales. La hipertensión arterial (HTA), definida como la elevación constante de la presión arterial, refleja una disrupción en el delicado equilibrio que mantienen los diversos factores mencionados.
En la enorme mayoría de los casos (95%), la HTA no depende de una causa única, sino que es de origen poligénico y multifactorial. Solo 5% de los casos de HTA tiene como causa la mutación de un gen único, que se transmite en la familia siguiendo un modelo mendeliano.
Debido a su origen poligénico y multifactorial, la PA constituye una característica fenotípica cuantitativa que se distribuye en la población siguiendo la curva de Gauss, y el límite entre la normotensión y la hipertensión se establece arbitrariamente a partir de criterios que pueden variar- y de hecho han variado- a lo largo del tiempo.
En esta revisión nos ocuparemos de la influencia del genoma y el epigenoma en la determinación de la presión arterial, es decir, de los genes que intervienen en la fisiopatología de la hipertensión arterial, y de los factores epigenéticos, que representan el puente a través del cual los factores ambientales interactúan con los genes y pueden modular la expresión génica.
La influencia del genoma en la regulación de la PA explica la agregación familiar que se observa en la HTA, aunque la cuantificación de la importancia relativa de cada uno de los dos grupos de factores, genéticos y ambientales, es aún materia de discusión y las cifras difieren en los diferentes trabajos publicados. En general, analizando los datos tanto de estudios poblacionales como de la comparación entre gemelos monocigóticos y dicigóticos, la heredabilidad de la HTA se estima entre 30 y 50% (1).
GENES RELACIONADOS CON LA HTA PRIMARIA
Dada la íntima relación clínica de HTA primaria con otras patologías -como diabetes, aterosclerosis e hiperlipidemia, todas ellas componentes del sindrome metabólico-, no llama la atención que los mismos genes que intervienen en estos fenotipos estén también implicados en HTA.
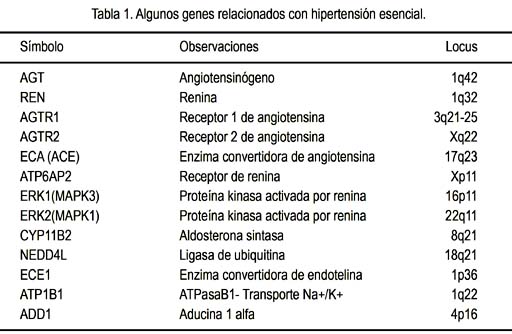
Hasta el momento se ha identificado unos 150 loci (lugares) cromosómicos que alojan genes directa o indirectamente relacionados con HTA. Estos genes codifican proteínas que influencian en cualquiera de los factores fisiopatológicos determinantes de HTA: contractibilidad miocárdica, volemia y resistencia vascular. Los genes candidatos más importantes son probablemente los relacionados al sistema renina-angiotensina-aldosterona (SRAA), pero también se considera los relacionados a la síntesis y metabolización de los esteroides adrenales y los que afectan el tono vascular, el transporte iónico y el manejo renal del sodio, entre otros. Algunos de estos genes y sus respectivos loci se muestran en la tabla. Ciertas variantes (polimorfismos) de estos genes se encuentran con mayor frecuencia en la población hipertensa que en la normotensa; por eso, son considerados alelos de riesgo. Mencionaremos solo algunos ejemplos:
El gen del angiotensinógeno se ubica en el cromosoma 1 y es activo especialmente en las células del hígado, encargadas de la síntesis de angiotensinógeno, la cual está en cierta medida influenciada por hormonas como los estrógenos, los glucocorticoides y las hormonas tiroideas, entre otras. El gen tiene una determinada secuencia de nucleótidos, pero existen algunas variantes (polimorfismos) de esta secuencia presentes en un porcentaje variable de la población, y que resultan en un polipéptido levemente distinto del más común; las variantes más comunes son M235T y T174M, que significa en el caso del M235T que en el aminoácido 235 una metionina ha sido reemplazada por treonina y en el segundo caso, T174M, que en el aminoácido 174 se encuentra metionina en vez de treonina. Ambas variantes se hallan en diferente proporción en diversas poblaciones y se relacionan con mayor frecuencia de HTA en quienes las portan, especialmente para los que son homocigotas para alguno de estos polimorfismos o quienes portan uno o dos alelos que combinan ambas variantes (2-4).
El gen que codifica renina está también en el cromosoma 1, en un locus diferente al del angiotensinógeno, y ejerce su acción en las células yuxtaglomerulares del riñón, encargadas de secretar esta enzima, la cual, al clivar el angiotensinógeno para formar angiotensina I, inicia la cascada de eventos del SRAA, que culmina en la formación de aldosterona, con la consecuente vasoconstricción y elevación de la PA. En este sistema interviene también el gen del receptor de renina, llamado ATP6AP2, descubierto en el año 2002 (5), que se ubica en el cromosoma X. La renina unida a su receptor induce un ritmo de formación de angiotensina I cuatro veces mayor que el que induce la renina soluble. Al mismo tiempo, la estimulación de la renina activa los genes ERK1 (MAPK3) y ERK2 (MAPK1), que se relacionan con obesidad y con hipertrofia cardiaca, entre otras cosas (6,7). Otras mutaciones del gen receptor de renina ocasionan fenotipos diversos no relacionados con HTA (8). Luego de la renina, la siguiente enzima que participa en el sistema es la convertidora de angiotensina, ECA, que convierte la angiotensina I en el péptido activo, angiotensina II; el gen ECA tiene dos posibles variantes: I (inserción) o D (deleción), referidos a la presencia o ausencia de un fragmento de 287 pares de bases en el intrón 16 del gen. La presencia del alelo D se relaciona con mayor actividad plasmática de la ECA, especialmente en homocigotos DD (9) y también con menor respuesta hipertensiva al metoprolol, en comparación con los homocigotos para el alelo con inserción (genotipo II), al menos en ciertas poblaciones (10). No es claro cómo actúa este polimorfismo, ya que por estar situado en un intrón no se transcribe y por lo tanto el polipéptido resultante no debe ser diferente en uno y otro caso; se especula que el polimorfismo I/D podría ser solo un marcador ligado a la verdadera variante del gen responsable de los mencionados efectos fenotípicos; posteriores estudios contribuirán a aclarar este punto, así como el hecho que estas coronaria y accidente cerebrovascular (9). La angiotensina II interactúa con dos tipos de receptores de la superficie celular, tipo 1 y tipo 2, codificados por los genes AGTR1 y AGTR2, de los cuales el primero es el más importante, ya que su expresión aumentada induce hipertrofia del miocardio e HTA (11); desde el punto de vista farmacológico, los antagonistas del receptor 1, como el losartán, son efectivos en el tratamiento de la HTA dependiente de angiotensina II.
También interviene el gen de la aldosterona sintasa (CYP11B2), que cuando presenta el polimorfismo -344C/T confiere aumento de riesgo para infarto cerebral e hipertensión (12,13) y los genes HSD3B1 and HSD3B2, que codifican enzimas necesarias para la síntesis de esteroides hormonales, incluida la aldosterona y cuyos polimorfismos influyen en el riesgo de HTA (14).
El ejemplo ilustra la compleja relación entre los genes y nos recuerda que para la correcta interpretación de hallazgos genómicos es importante tomar en cuenta que ninguno de los genes involucrados en la regulación de la PA tiene un valor determinante ni actúa aisladamente; cada uno de ellos tiene un efecto poco importante, pero es la suma de alelos de riesgo, o la manera como estos se combinan, lo que influye en el fenotipo final. Por ejemplo, la presencia del alelo M235T del angiotensinógeno, que se encuentra en 40% de la población, confiere un riesgo relativo de sufrir IMA de aproximadamente 2 y el alelo D de la ECA de 1.8. Sin embargo, la presencia de ambos alelos no es aditiva, sino que su combinación ejerce un efecto multiplicador, ya que si alguien presenta ambos alelos de riesgo, su riesgo de IMA es de 11 (9).
El gen ADD1, situado en el cromosoma 4p, codifica una proteína llamada aducina 1, que se encuentra en el citoesqueleto de membrana y que favorece la unión entre la espectrina y la actina. Debido a su interacción entre los filamentos de actina y de espectrina tiene una importante función en la arquitectura de la membrana y, potencialmente, sobre la actividad de ciertos canales, en particular el cotransporte de Na-K-Cl y la Na-K-ATPasa. El polimorfismo G460W del gen de la aducina1 es más frecuente en los hipertensos que en los normotensos y parece predisponer a una sensibilidad particular a la sal y a la hipertensión. La influencia de otros genes relacionados, la aducina 2 y 3 (genes ADD2 y ADD3), no es aún clara (15). Debemos recordar que los genes que contribuyen a la HTA primaria son loci cuantitativos (QTL), identificados a través de estudios de asociación y estudios en modelos animales y cuyos resultados pueden diferir en diferentes poblaciones (16).
FORMAS MONOGÉNICAS DE HIPERTENSIÓN ARTERIAL
Todos los cuadros en que un solo gen es responsable de la HTA son raros. Debe sospecharse esta etiología cuando la genealogía muestra un patrón de herencia definido, sea dominante o recesivo; generalmente se trata de HTA severa y muchas veces de inicio temprano, incluso en la niñez o adolescencia.
Hiperaldosteronismo familiar tipo 1
También llamado hiperaldosteronismo remediable con glucocorticoides (GRA) o hiperaldosteronismo suprimible con glucocorticoides (GSH), es causado por un defecto genético puntual, la fusión de dos genes contiguos, el CYP11B2 y CYP11B1, que se encuentran en el brazo largo del cromosoma 8. Este gen híbrido es responsable de un cuadro de hipertensión arterial por hiperaldosteronismo que, como su nombre lo indica, puede ser tratado con éxito con dexametasona. Es una forma muy rara de hiperaldosteronismo primario, que se manifiesta generalmente en la edad adulta. La herencia es autosómica dominante, aunque con penetrancia incompleta, es decir que algunos portadores no manifiestan el fenotipo (17). En las familias con personas afectadas es posible detectar portadores con un examen directo del gen, que por ahora solo se hace en cuatro laboratorios en el mundo, y tomar medidas preventivas en quienes portan esta mutación.
Hiperaldosteronismo familiar tipo 2
Tiene los mismos síntomas que el tipo 1, pero no responde al tratamiento con dexametasona. No se conoce aún el gen causante, pero los estudios de ligamiento indican que estaría ubicado en el brazo corto del cromosoma 7, región 7p22 (18).
Síndrome de exceso aparente de mineralocorticoides (AME)
Causado por la mutación del gen HD11B2 que codifica la enzima cortisol 11-betacetoreductasa, que convierte el cortisol a cortisona (19). Al faltar la enzima, ocurre el bloqueo de esta vía, causando gran elevación de cortisol, el cual, por su abundancia, pasa a reemplazar a la aldosterona en su función de regular al receptor de mineralocorticoides, que resulta sobre-estimulado. El paciente tiene todos los síntomas de hiperaldosteronismo, con hipertensión e hipokalemia, que empiezan desde la infancia; el tratamiento con dexametasona para regular la secreción de cortisol, es efectivo. La herencia es autosómica recesiva.
Síndrome de Liddle
Llamado también pseudoaldosteronismo, presenta síntomas similares al síndrome de exceso aparente de mineralocorticoides (AME), pero sin respuesta efectiva a dexametasona. Hay hipertensión con hipokalemia, debido a una disfunción del canal epitelial renal de sodio, falla ocasionada por mutaciones de los genes que codifican la subunidad beta o gamma de este canal (SCNN1B y SCNN1G, respectivamente), ambos situados en el locus cromosómico 16p13-12, y para los cuales existe posibilidad de diagnóstico molecular. Otros genes que interactúan con los mencionados y cuyas mutaciones también causan el síndrome de Liddle son el NEDD4, NEDD4L y NR3C2. La herencia de este cuadro es autosómica dominante.
El pseudohipoaldosteronismo tipo 1 se debe a mutaciones raras que causan ganancia de función del gen receptor de mineralocorticoides NR3C2. El cuadro es de hipertensión de inicio temprano y con gran exacerbación en la gestación, con las complicaciones de parto prematuro y otras. La herencia es autosómica dominante (20). Investigadores de la Universidad de Leiden han encontrado recientemente que una variante común de este mismo gen, el polimorfismo c.-2G>C, es un alelo de riesgo para hipertensión esencial, asociado con mayor actividad del eje renina-angiotensina-aldosterona y aumento de presión arterial (21).
FACTORES EPIGENÉTICOS EN HIPERTENSIÓN ARTERIAL
El genoma humano haploide está compuesto de tres mil millones de pares de bases, divididos en 23 moléculas de ADN, que forman los cromosomas y que contienen unos 25 000 genes. Estos genes representan únicamente el 3% del genoma; el resto son secuencias no codificantes, cuya función es aún desconocida.
El genoma es idéntico en todas las células y potencialmente todos los genes podrían expresarse (transcribirse y formar ARN mensajero para culminar el proceso con la formación de un polipéptido) en cada célula y tejido. Pero, sabemos que esto no es así, ya que hay mecanismos, llamados epigenéticos, que silencian o activan genes, permitiendo la adaptación al entorno, uno de los procesos regulatorios fundamentales de los seres vivos, que depende en parte de la existencia de una gran diversidad de tipos celulares que proporcionan la capacidad de adaptación a los cambios ambientales. Actualmente se admite que, además del código genético, existe otro código que, independientemente de la secuencia del gen, determina la apertura o cierre de la cromatina para exponer o no una determinada región del ADN, permitiendo su transcripción. Este es el código epigenético, constituido por un sistema de moléculas unidas al complejo ADN/histonas, que a diferencia del inmutable código genético, es dinámico, flexible y modificable dependiendo de cambios químicos realizados sobre el ADN y/o las histonas, que a su vez son influidos por factores ambientales. Los genes se expresan o no dependiendo de ciertas condiciones bioquímicas, como la metilación del ADN, la acetilación de las histonas y otras.
Las alteraciones epigenéticas están implicadas en numerosas enfermedades comunes, incluyendo la hipertensión arterial (1). En la aterosclerosis, por ejemplo, la formación de la placa de ateroma se debe a una susceptibilidad genética, a una dieta rica en grasas y a la existencia de un patrón epigenético de expresión de genes relacionados con el metabolismo de las grasas. La aparente relación entre malnutrición materna, peso bajo de nacimiento y mayor probabilidad de presentar HTA en la edad adulta estaría condicionado por factores epigenéticos (22,23). Se postula que el vínculo entre dos períodos de desarrollo diferentes, como son el nacimiento y la adultez, serían los cambios en la regulación de ciertos genes, inducidos por factores ambientales, que llevarían finalmente a una susceptibilidad aumentada para ciertas enfermedades crónicas del adulto. Experimentos animales apoyan esta hipótesis (1). La preeclampsia, con su componente hipertensivo, también tiene una importante influencia epigenética (24).
PERSPECTIVAS
Es claro que, salvo en casos excepcionales, no hay un gen único o algunos genes que permitan explicar, por sí mismos la hipertensión arterial. Los experimentos con animales transgénicos, los estudios de asociación genómica amplia (GWA) y los de ligamiento revelan numerosos loci implicados y una multitud de genes de susceptibilidad, cuyo efecto fenotípico es manifiesto únicamente mediante interacción entre ellos y con el medio ambiente (16,25). Además, los genes implicados en HTA pueden ser diferentes según el origen étnico de la población estudiada (10). Todo esto impide que actualmente haya análisis genéticos realmente útiles para diagnosticar o predecir el riesgo personal o familiar de desarrollar hipertensión (25).
Por la misma razón, la farmacogenómica y la fármacogenética no son parte aún del arsenal terapéutico relacionado con HTA, pero se espera que lo sean conforme se vaya ampliando el rango de genes identificados y su mecanismo de acción. Las asociaciones mencionadas en párrafos anteriores entre nivel de ECA y polimorfismos del gen que lo codifica hicieron pensar que el análisis genético podría ser útil para correlacionar con la respuesta a inhibidores de ECA, pero en la práctica eso no ha podido ser replicado; lo mismo ha sucedido con la relación entre el polimorfismo Gly460Trp del gen ADD1 y la efectividad de los diuréticos tiazídicos en el control de la PA. Sin embargo, las investigaciones continúan y reciente mente se ha encontrado que cuando el mencionado polimorfismo de ADD1 se combina con otro del gen NEDD4L (el mismo implicado en síndrome de Liddle) sí hay una respuesta efectiva a tiazidas, lo que prueba una vez más la complejidad genética involucrada en la respuesta a fármacos. Igualmente, están siendo estudiados los polimorfismos del gen ADRB1, que codifica el receptor adrenérgico beta1, en relación a la respuesta a los beta bloqueadores, con resultados que parecen ser promisorios (26). Los análisis de ciertas variantes del citocromo P450 implicadas en el metabolismo de drogas ya están disponibles y son utilizados en otras especialidades médicas, pero no son realmente útiles en caso de hipertensión (27). Se espera que en la década que comienza sea más accesible el análisis del genoma individual y se tenga más clara la relación entre ciertas características genómicas (SNPs) y la respuesta a determinados fármacos, de modo que el médico pueda seleccionar la medicación más efectiva para su paciente.
Por otra parte, es indudable que la medicina está entrando en una etapa en que el paciente está siendo empoderado para asumir sus propias decisiones en muchos aspectos del cuidado de su salud. Y, a pesar de lo expuesto en los párrafos anteriores respecto a la complejidad de la interpretación de hallazgos genómicos, hay una gran oferta de pruebas ofrecidas directamente al consumidor, en este caso el paciente, obviando la intervención del médico. Estas pruebas no están aún clínicamente validadas, pero su accesibilidad, unida a eficientes tácticas de mercadeo a través de internet, están popularizando su uso, con el peligro de que el paciente tome decisiones inadecuadas basado únicamente en ciertos análisis y sin tomar en cuenta todos los múltiples factores que intervienen en la génesis de un proceso patológico, en este caso la HTA. Sin embargo, la revolución genética está presente y se trata de una realidad insoslayable. En un futuro cercano, la tarea del médico probablemente será la de de interpretar las pruebas genéticas que su paciente solicitó directamente y, en base a esa información, indicar medidas preventivas y tratamiento (28).
Los organismos regulatorios están ya preocupándose de evaluar rigurosamente la validación clínica y la utilidad de las pruebas genómicas ofrecidas (27), lo cual permitirá trabajar conjuntamente con los clínicos e investigadores para beneficiar al paciente de una medicina personalizada que, bien aplicada, tiene un enorme potencial en la mejora de la salud.
REFERENCIAS BIBLIOGRÁFICAS
1. Kunes J, Zicha J. The interaction of genetic and environmental factors in the etiology of hyper tension. Physiol Res. 2009;58 Suppl 2:S33-41.
2. Brand E, Chatelain N, Paillard F, Tiret L, Visvikis S, Lathrop M, Soubrier F, Demenais F. Detection of putative functional angiotensinogen (AGT) gene variants controlling plasma AGT levels by combined segregation-linkage analysis. Europ J Hum Genet. 2002;10:715-23.
3. Markovic D, Tang X, Guruju M, Levenstien MA, Hoh J, Kumar A, Ott J. Association of angiotensinogen gene polymorphisms with essential hypertension in African-Americans and Caucasians. Hum Hered. 2005;60:89-96.
4. Nakajima T, Wooding S, Sakagami T, Emi M, Tokunaga K, Tamiya G, Ishigami T, Umemura S, Munkhbat B, Jin F, Guan-jun J, Hayasaka I, Ishida T, Saitou N, Pavelka K, Lalouel J-M, Jorde LB, Inoue I. Natural selection and population history in the human angiotensinogen gene (AGT): 736 complete AGT sequences in chromosomes from around the world. Am J Hum Genet. 2004;74:898-916.
5. Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer J-D. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417-27.
6. Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nature Med. 2009;15:75-83.
7. Imai J, Katagiri H, Yamada T, Ishigaki Y, Suzuki T, Kudo H, Uno K, Hasegawa Y, Gao J, Kaneko K, Ishihara H, Niijima A, Nakazato M, Asano T, Minokoshi Y, Oka Y. Regulation of pancreatic beta cell mass by neuronal signals from the liver. Science. 2008;322:1250-4.
8. Ramser J, Abidi FE, Burckle CA, Lenski C, Toriello H, Wen G, Lubs HA, Engert S, Stevenson RE, Meindl A, Schwartz CE, Nguyen G. A unique exonic splice enhancer mutation in a family with X-linked mental retardation and epilepsy points to a novel role of the renin receptor. Hum Molec Genet. 2005;14:1019-27.
9. Pirola CJ. Genética molecular de la hipertensión arterial esencial. Medicina (Buenos Aires). 2000;60:59-66.
10. Liu C, Li H, Qi Q, Lu L, Gan W, Loos RJ, Lin X. Common variants in or near FGF5, CYP17A1 and MTHFR genes are associated with blood pressure and hypertension in Chinese Hans. J Hypertens. 2010 Sep 16 [Publicación electrónica antes de la impresión].
11. Niu W, Qi Y. Association of the angiotensin II type I receptor gene +1166 A>C polymorphism with hypertension risk: evidence from a meta-analysis of 16474 subjects. Hypertens Res. 2010 Aug 12. [Publicación electrónica antes de la impresión].
12. Munshi A, Sharma V, Kaul S, Rajeshwar K, Babu MS, Shafi G, Anila AN, Balakrishna N, Alladi S, Jyothy A. Association of the -344C/T aldosterone synthase (CYP11B2) gene variant with hypertension and stroke. J Neurol Sci. 2010;296(1-2):34-8.
13. Sookoian S, Gianotti TF, González CD, Pirola CJ. Association of the C-344T aldosterone synthase gene variant with essential hypertension: a metaanalysis. J Hypertens. 2007;25(1):5-13.
14. Shimodaira M, Nakayama T, Sato N, Aoi N, Sato M, Izumi Y, Soma M, Matsumoto K. Association of HSD3B1 and HSD3B2 gene polymorphisms with essential hypertension, aldosterone level, and left ventricular structure. Eur J Endocrinol. 2010;163(4):671-80.
15. Lanzani C, Citterio L, Jankaricova M, Sciarrone MT, Barlassina C, Fattori S, Messaggio E, Di Serio C, Zagato L, Cusi D, Hamlyn JM, Stella A, Bianchi G, Manunta P. Role of the adducin family genes in human essential hypertension.
J Hypertens. 2005;23:543-9.16. Deng AY. Genetic basis of polygenic hypertension. Hum Mol Genet. 2007 Oct 15;16(R2): R195R202.
17. Mulatero P, Di Cella SM, Williams TA, Milan A, Mengozzi G, Chiandussi L, Gomez-Sanchez CE, Veglio F. Glucocorticoid remediable aldosteronism: low morbidity and mortality in a four-generation Italian pedigree. J Clin Endocr Metab. 2002;87:3187-91.
18. Lafferty AR, Torpy DJ, Stowasser M, Taymans SE, Lin JP, Huggard P, Gordon RD, Stratakis CA. A novel genetic locus for low renin hypertension: familial hyperaldosteronism type II maps to chromosome 7 (7p22). J Med Genet. 2000;37(11):831-5.
19. Kamide K, Kokubo Y, Hanada H, Nagura J, Yang J, Takiuchi S, Tanaka C, Banno M, Miwa Y, Yoshii M, Matayoshi T, Yasuda H, Horio T, Okayama A, Tomoike H, Kawano Y, Miyata T. Genetic variations of HSD11B2 in hypertensive patients and in the general population, six rare missense/frameshift mutations. Hypertens Res. 2006;29(4):243-52.
20. Geller DS, Farhi A, Pinkerton N, Fradley M, Moritz M, Spitzer A, Meinke G, Tsai FTF, Sigler PB, Lifton RP.Activatingmineralocorticoid receptormutation in hypertension exacerbated by pregnancy. Science. 2000;289(5476):119-23.
21. van Leeuwen N, Caprio M, Blaya C, Fumeron F, Sartorato P, Ronconi V, Giacchetti G, Mantero F, Fernandes-Rosa FL, Simian C, Peyrard S, Zitman FG, Penninx BW, de Kloet ER, Azizi M, Jeunemaitre X, Derijk RH, Zennaro MC. The functional c.2G>C variant of the mineralocorticoid receptor modulates blood pressure, renin, and aldosterone levels. Hypertension. 2010 Sep 20 [Publicación electrónica antes de la impresión].
22. Barker DJ, Osmond C, Forsen TJ, Kajantie E, Eriksson JG. Maternal and social origins of hypertension. Hypertension. 2007;50(3):565-71.
23. Eriksson JG, Forsén TJ, Kajantie E, Osmond C, Barker DJ. Childhood growth and hypertension in later life. Hypertension. 2007;49(6):1415-21.
24. Chelbi ST, Vaiman D. Genetic and epigenetic factors contribute to the onset of preeclampsia. Mol Cell Endocrinol. 2008;282(1-2):120-9.
25. Rafiq S, Anand S, Rober ts R. Genome-wide association studies of hyper tension: have they been fruitful? J Cardiovasc Transl Res. 2010;3(3):189-96.
26. Johnson JA. Pharmacogenomics of antihypertensive drugs: past, present and future. Pharmacogenomics. 2010;11(4):487-91.
27. Annes JP, Giovanni MA, Murray MF. Risks of presymptomatic direct-to-consumer genetic testing. N Engl J Med. 2010;363(12):1100-1.
28. Evans JP, Dale D, Formous C. Preparing for a consumer-driven genomic age. N Engl J Med. 2010;363(12):1099-103.
Correspondencia:
Dra. María Isabel Quiroga de Michelena
mqmichelena@genetica.com.pe

