










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkAnales de la Facultad de Medicina
versão impressa ISSN 1025-5583
An. Fac. med. v.72 n.1 Lima jan./mar. 2011
ARTÍCULO DE REVISIÓN
Disfunción tiroidea inducida por amiodarona en la práctica clínica
Thyroid dysfunction induced by amiodarone in clinical practice
José Luis Paz-Ibarra
Servicio de Endocrinología, Hospital Nacional Edgardo Rebagliati Martins, EsSalud.
Facultad de Medicina, Universidad Nacional Mayor de San Marcos.
RESUMEN
La amiodarona (AMD) es una droga antiarrítmica potente (clase III) usada en la práctica clínica para la profilaxis y el tratamiento de muchos disturbios del ritmo cardiaco, desde la fibrilación auricular paroxística hasta las taquiarritmias ventriculares que amenazan la vida. Frecuentemente causa cambios en las pruebas de función tiroidea principalmente relacionados a la inhibición de la actividad de la 5'-deiodinasa, resultando en una disminución de la generación de T3 desde T4 y el consecuente incremento en la producción de T3 reversa y una disminución de su aclaramiento. En 14 a 18% de pacientes tratados con AMD hay una disfunción tiroidea manifiesta, ya sea tirotoxicosis inducida por amiodarona (TIA) o hipotiroidismo inducido por amiodarona (HIA). Tanto TIA como HIA pueden desarrollarse en glándulas aparentemente normales o en glándulas con anormalidades preexistentes clínicamente silentes. La TIA está primariamente relacionada a la síntesis de hormonas tiroideas inducida por el exceso de yodo en una glándula tiroidea anormal (TIA tipo 1) o a una tiroiditis destructiva relacionada a la amiodarona (TIA tipo 2), aunque frecuentemente ocurren formas mixtas. La tiroiditis de Hashimoto preexistente es un factor de riesgo definido para la ocurrencia de HIA. La patogenia del HIA es la falla para escapar del efecto agudo de Wolff-Chaikoff inducido por el yodo, debido a los defectos en la hormonogénesis tiroidea y, en pacientes con pruebas de autoanticuerpos tiroideos positivos, para tiroiditis de Hashimoto concomitante. La TIA es más común en zonas deficientes de yodo mientras que el HIA es usualmente visto en zonas suficientes en yodo. En contraste al HIA, la TIA es una condición difícil de diagnosticar y tratar, y usualmente se recomienda la descontinuación de la amiodarona. En esta revisión se analiza, de acuerdo a los datos actuales, las alteraciones en las pruebas de función tiroidea vistas en pacientes eutirodeos bajo tratamiento con AMD así como la epidemiología y opciones de tratamiento disponibles para ambos tipo de disfunción tiroidea inducida por amiodarona, TIA e HIA.
Palabras clave: Amiodarona, tirotoxicosis inducida por amiodarona, hipotiroidismo inducido por amiodarona.
ABSTRACT
Amiodarone is a potent class III anti-arrhythmic drug used in clinical practice for the prophylaxis and treatment of many cardiac rhythm disturbances, ranging from paroxismal atrial fibrillation to life threatening ventricular tachyarrhythmias. Amiodarone often causes changes in thyroid function tests mainly related to inhibition of 5'-deiodinase activity resulting in decrease in the generation of T3 from T4 with consequent increase in rT3 production and decrease in its clearance. In 14-18% of amiodarone-treated patients, there is overt thyroid dysfunction, either amiodarone-induced thyrotoxicosis (AIT) or amiodarone-induced hypothyroidism (AIH). Both AIT and AIH may develop either in apparently normal thyroid glands or in glands with preexisting clinically silent abnormalities. AIT is primarily related to excess iodine-induced thyroid hormone synthesis in an abnormal thyroid gland (type I AIT) or to amiodarone-related destructive thyroiditis (type II AIT). AIH pathogenesis is related to escape failure from the acute Wolff-Chaikoff effect due to defects in thyroid hormonogenesis or, in patients with positive thyroid autoantibody test, to concomitant Hashimoto's thyroiditis. AIT is more common in iodine-deficient regions of the world, whereas AIH is usually seen in iodine-sufficient areas. In contrast to AIH, AIT is a condition difficult to diagnose and treat, and discontinuation of amiodarone is usually recommended. In this review discusses, according to data from current literature, alterations in thyroid laboratory tests seen in euthyroid patients under treatment with amiodarone and the epidemiology and treatment options available of amiodarone-induced thyroid dysfunctions (AIT and AIH).
Keywords: Amiodarone, amiodarone-induced thyrotoxicosis, amiodarone-induced hypothyroidism.
INTRODUCCIÓN
La amiodarona (AMD) es un fármaco antiarrítmico de clase III; junto al sotalol y dofetilida, retarda los impulsos eléctricos del corazón, bloqueando los canales de potasio miocárdicos, además de poseer algunas propiedades betabloqueantes (1). Actualmente, es uno de los más usados para la profilaxis y el control de las taquiarritmias más frecuentes, indicándose su administración por vía EV inicialmente y luego por vía oral, siendo mantenida por varias semanas e incluso meses, según sea la indicación terapéutica.
Como todo fármaco, no está exento de efectos adversos y a nivel cardiovascular ha sido asociado a un aumento de la mortalidad en pacientes con insuficiencia cardiaca por su efecto inotrópico negativo y prolongación del intervalo QT; además, su uso se asocia a diferentes efectos adversos, siendo los órganos más afectados la córnea, pulmones, hígado, piel y la glándula tiroides (1,2). A nivel pulmonar, se ha encontrado neumonitis por hipersensibilidad o por fosfolipoidosis diseminada y fibrosis pulmonar; a nivel hepático, se ha hallado elevación de transaminasas y hepatitis aguda tipo alcohólica; a nivel cutáneo existe fotosensibilidad y en algunos pacientes tinte azul-grisáceo; a nivel ocular microdepósitos corneales, fotofobia y visión borrosa; y a nivel del sistema nervioso, ataxia, temblor, debilidad muscular y neuropatía periférica. Sin embargo, algunos han encontrado algunos efectos beneficiosos como la elevación del colesterol de alta densidad (HDL) con el uso prolongado de este medicamento (4).
A nivel de tiroides, la AMD puede alterar el metabolismo de las hormonas tiroideas, resultando en alteraciones en las pruebas de función tiroidea en individuos eutirodeos, y puede causar tanto hipotiroidismo como hipertiroidismo, lo que se conoce como disfunción tiroidea inducida por amiodarona (DTIA) (3-5).
CARACTERÍSTICAS DE LA AMIODARONA
Es un derivado de benzofurano iodado y posee una semejanza estructural significativa a las hormonas tiroideas (HT). Cada molécula de AMD contiene dos átomos de I (yodo), los cuales constituyen el 37,5% de su masa (figura 1).
Así, una ingesta de 200 mg/día de AMD significa la ingesta de 75 mg de yodo orgánico cada día. Diariamente se deiodiniza el 10% de la molécula; por tanto, su deiodinización subsecuente resulta en la liberación diaria de 6 a 7 mg de yodo libre a la circulación. Farmacológicamente, la AMD tiene una vida media muy larga (aproximadamente 100 días), principalmente debido a su almacenamiento en el tejido adiposo. Por ello, el exceso de yodo es aclarado lentamente durante meses y sus efectos tóxicos pueden persistir o incluso ocurrir después de descontinuarla.
La amiodarona es metabolizada por desalquilación a desetilAMD (DEA), un metabolito activo (5-7).
AMIODARONA Y TIROIDES
Aunque la mayoría de los efectos adversos de la AMD sobre varios órganos se deben al depósito de la droga en el parénquima, sus efectos sobre la glándula tiroides y el metabolismo de las hormonas tiroideas (HT) son únicos (figura 2).
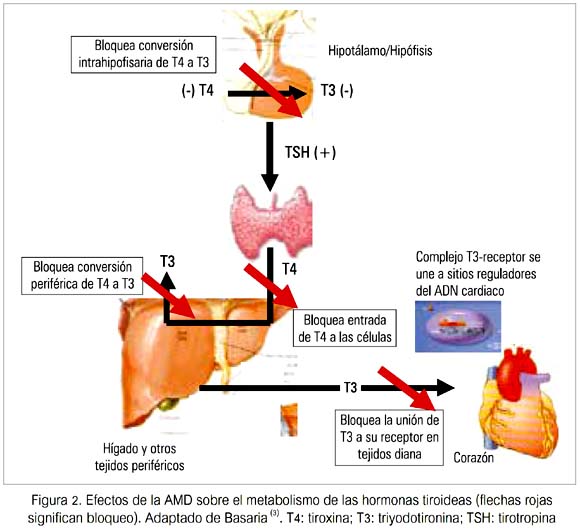
Estos efectos pueden ser divididos en efectos intrínsecos del compuesto sobre la glándula tiroides y el metabolismo de las HT (tabla 1) y los efectos inducidos por el yodo (debido únicamente a los efectos farmacológicos de una sobrecarga de yodo) (tabla 2).


CAMBIOS EN LAS HORMONAS TIROIDEAS INDUCIDOS POR AMD EN SUJETOS EUTIROIDEOS
La mayoría de pacientes que reciben AMD permanece eutiroidea. En un estudio prospectivo en los EE.UU., el seguimiento de 76 pacientes eutiroideos que recibieron AMD por 16 meses mostró que 89% permaneció eutirodeo (3). Debido a que la tiroides está expuesta a una carga extraordinaria de yodo, se desarrollan importantes ajustes en el manejo del yodo y el metabolismo de las hormonas tiroideas, en orden de mantener su función normal, lo cual se refleja en las pruebas de función tiroidea (PFT) (figura 3).
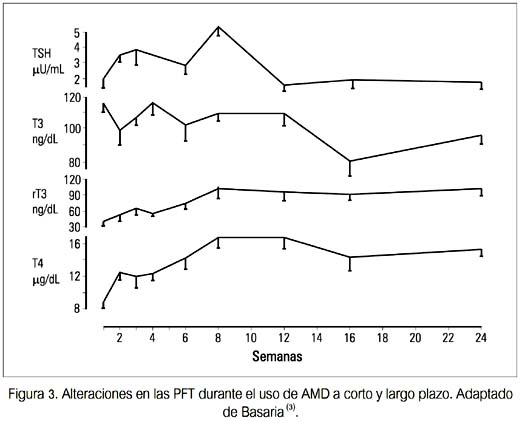
Estas alteraciones pueden dividirse en dos fases, agudas (<3 meses) y crónicas (>3 meses) (tabla 3).

HIPOTIROIDISMO INDUCIDO POR AMD
Factores de riesgo
Ingesta de I (yodo):
El hipotiroidismo inducido por AMD (HIA) es más prevalente en áreas suficientes de yodo. Así en Toscana - Italia (zona deficiente de I) es de 5%; mientras que en Worcester - Massachussets (zona suficiente de I) es de 22% (3,10,11).
Tiroiditis autoinmune:
Es el factor de riesgo más común para el desarrollo y persistencia de HIA; es más frecuente en mujeres que en varones. El 70% de pacientes con HIA persistente tenía AcTPO (+), mientras que 90% de pacientes sin AcTPO tuvo HIA transitorio que se resolvió dentro de los 2 a 4 meses de suspendida la AMD. El 71% de pacientes con T. de Hashimoto (TH) subclínico desarrolló HIA, comparado con ninguno en los pacientes sin TH.
La presencia de AcTPO confirió un RR de HIA de 7,3; el sexo femenino confirió un RR de 7,9. Ambos factores confieren un RR de 13,5. Sin embargo, al momento actual queda sin responder la siguiente interrogante: ¿El uso de AMD induce la formación de anticuerpos antitiroideos?
TSH basal:
La elevación basal de TSH antes del uso de AMD ha mostrado ser un factor de riesgo para HIA, probablemente reflejando la ETAI subyacente. En un estudio, el promedio de TSH en los que desarrollaron HIA fue 5,6 mU/L versus 3,9 mU/L en los sujetos que permanecieron eutiroideos (3).
Patogénesis
La incapacidad para escapar al efecto Wolff-Chaikoff puede ser el mecanismo más probable; por otro lado, es posible que la liberación de auto-antígenos desde la tiroides lesionada pueda llevar a la potenciación de la autoinmunidad, acelerando así el curso natural de la TH (3,4)
Clínica y laboratorio
El HIA típicamente ocurre entre 6 a 12 meses de tratamiento con AMD. No hay diferencias en las manifestaciones clínicas entre pacientes con HIA comparados con aquellos con hipotiroidismo debido a otras causas. Los síntomas son vagos e incluye piel seca, letargia, intolerancia al frío y capacidad de concentración alterada. Se ha comunicado un caso de coma mixedematoso en un paciente que tomaba AMD. El bocio es raramente visto en asociación con HIA (12)
Los hallazgos de laboratorio en los pacientes con HIA son similares a los pacientes con hipotiroidismo espontáneo, es decir, TSH elevado y T4L bajo; sin embargo, los niveles elevados de TSH no son muy útiles los tres primeros meses de tratamiento con AMD, debido a que ellos están elevados en este estadio.
Los valores >20 mIU/L son comúnmente vistos en pacientes con HIA, pero son raros en pacientes eutiroideos en el curso del tratamiento (13).
Tratamiento
El reemplazo con levotiroxina (LT4) es la terapia de elección. Si la AMD no puede ser descontinuada, LT4 restaura el eutiroidismo bioquímico, con resolución de síntomas. La terapia con LT4 se inicia con dosis bajas de 25 a 50 ug/d, y se va incrementando lentamente hasta que la TSH retorne a lo normal (14). La dosis de LT4 necesaria para normalizar TSH puede ser mayor en los pacientes con HIA comparados con los pacientes portadores de hipotiroidismos convencionales; posiblemente como resultado de la producción disminuida de T3 intrahipofisiaria, debido a la inhibición de la 5 deiodinasa tipo 2.
Un estudio mostró que la dosis media de LT4 necesaria para normalizar TSH en pacientes con HIA fue 256 µg/d, comparado con 136 µg/d en pacientes con hipotiroidismo espontáneo (15).
Si la AMD puede ser descontinuada, el retorno espontáneo al eutirodismo ocurre dentro de 2 a 4 meses en pacientes sin TH subyacente. La LT4 puede ser administrada si el paciente está sintomático (figura 4).
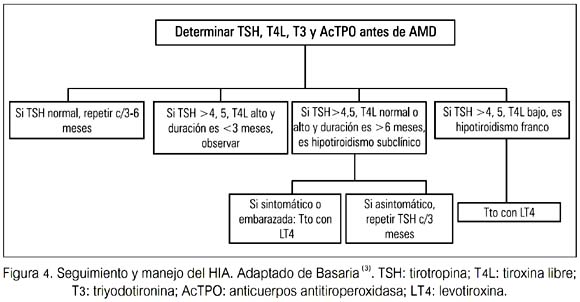
Debido a que el hipotiroidismo resuelve en pacientes sin autoinmunidad tiroidea subyacente, los pacientes deberían ser reevaluados después de 6 a 12 meses de terapia, para determinar la necesidad de continuar LT4. Los pacientes con Anticuerpos antitiroideos (AcAT) pueden desarrollar hipotiroidismo persistente, aún cuando la AMD haya sido descontinuada (16) (tabla 4).

TIROTOXICOSIS INDUCIDA POR AMD
La AMD ha llegado a ser una causa bien conocida y notoria de hipertiroidismo. Se ha descrito que la prevalencia de tirotoxicosis inducida por AMD (TIA) difiere entre poblaciones dependiendo de la ingesta de yodo. La diferencia en la prevalencia probablemente relaciona a la bien establecida tendencia de los individuos que viven en ambientes deficientes de yodo que llegan a ser hipertiroideos cuando son expuestos a yodo exógeno (suplemento nutricional o medicamentos que contienen yodo). A su vez, la deficiencia de yodo está ligada a la presencia de BMN no tóxico visto en sujetos yodo-deficientes, siendo más sensibles al yodo exógeno.
Tipos de TIA
Bartalena y colaboradores han categorizado la TIA en dos subtipos (17):
-
TIA tipo 1.
-
TIA tipo 2.
TIA tipo 1
Es definido como el hipertiroidismo inducido por yodo que se desarrolla en individuos quienes tienen una enfermedad tiroidea subyacente, y es debido a la síntesis y liberación incrementada de HT. Típicamente, ocurre en personas con BMN no tóxico preexistente o enfermedad de Graves latente subyacente, en quienes la exposición al yodo dispara el desarrollo del hipertiroidismo.
TIA tipo 2
Es una tiroiditis destructiva inducida por drogas, que ocurre en individuos sin enfermedad tiroidea subyacente. El hipertiroidismo es debido a la liberación de hormonas preformadas a la circulación, desde el epitelio folicular dañado.
En modelos in vitro de TIA se ha confirmado que AMD y DEA son tóxicos para las células tiroideas humanas y animales. Estudios histológicos de tiroides extirpadas de pacientes con TIA también han mostrado evidencia de daño folicular, infiltración histiocítica y fibrosis (figura 5) (18-20).
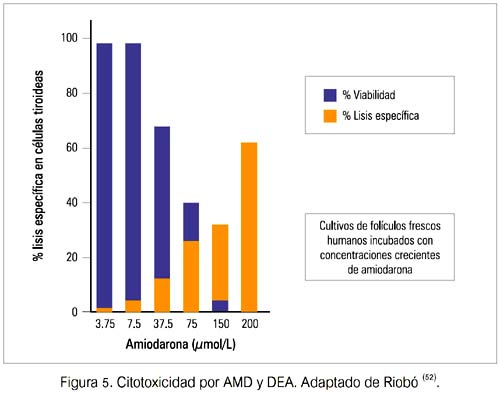
Como en otras formas de tiroiditis, la TIA puede ser seguida por una fase de hipotiroidismo transitorio antes de la recuperación total. Sin embargo, algunos pacientes con enfermedad tiroidea autoinmune (ETAI) subyacente pueden llegar a ser hipotiroideos permanentemente, luego de un episodio de TIA tipo 2.
Epidemiología
En contraste con la mayoría de enfermedades tiroideas, la TIA es más común en hombres. Esto probablemente es el resultado de una mayor frecuencia de arritmias y terapia con AMD en hombres. La prevalencia de TIA fue 9,6% y 2% en Tuscany y Worcester, respectivamente. De igual manera en un estudio holandés la prevalencia de TIA fue 12,1%.
Es difícil de estar seguro sobre la frecuencia relativa de los dos subtipos de TIA, porque probablemente varía según el estado de yodo-disponibilidad de una población. La TIA tipo 1 puede ser más común en las partes deficientes de yodo del mundo (Europa) y evidencias anecdóticas sugieren que la TIA tipo 2 es más común en las áreas suficientes de yodo del mundo (incluyendo los Estados Unidos y el Reino Unido). Además, existen probablemente formas mixtas de TIA, donde los individuos pueden tener las características de ambos subtipos de TIA (21,22).
Clínica
El cuadro clínico de TIA es similar en ambos tipos. En otros países, la función tiroidea es frecuentemente monitoreada en los pacientes que toman AMD, por lo que el diagnóstico es frecuentemente hecho a través del tamizaje, hecho que no se ha estandarizado en la práctica clínica en nuestro medio.
Generalmente el inicio es rápido, de manera que la función tiroidea puede ser normal incluso solo algunos meses antes del desarrollo de evidencia clínica o bioquímica de hipertiroidismo. Los pacientes ancianos pueden ser asintomáticos o simplemente presentar pérdida ponderal u otros síntomas inespecíficos. Debido a los efectos beta-bloqueantes de la AMD, las palpitaciones y la taquicardia están usualmente ausentes. En algunas ocasiones, la ocurrencia de novo de FA o empeoramiento de las arritmias, las cuales previamente se encontraban controladas, sugieren el diagnóstico.
Raramente la TIA tipo 2 presentará dolor tiroideo, fiebre y otros síntomas sistémicos. La orbitopatía está usualmente ausente. La glándula tiroides puede estar crecida y nodular en la TIA 1, mientras que en la TIA 2, la glándula está usualmente normal o mínimamente crecida.
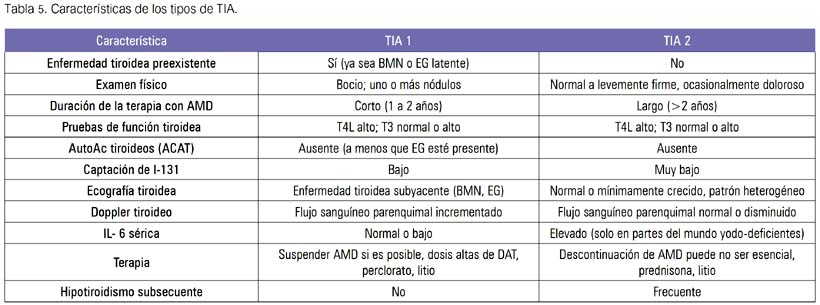
A nivel bioquímico existe aumento de T4 libre, TSH disminuida o suprimida y T3 aumentada, pero puede estar normal o en el límite inferior. La globulina ligadora de las hormonas sexuales (SHBG) está aumentada en la TIA y normal en los pacientes eutiroideos con hipertiroxinemia. Las características de ambos tipos de TIA son descritas en la tabla 5.
Tratamiento
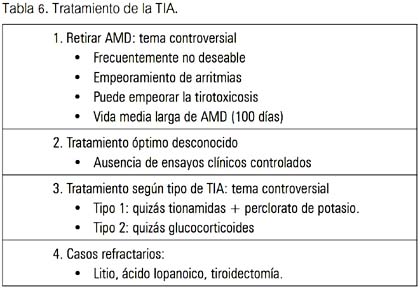
Debido a que la patogénesis de TIA no está completamente entendida, la terapia específica dirigida a la causa subyacente puede o no ser efectiva en cada caso. Además, los estudios realizados en la década de los 80 e inicios de los 90 no diferencian claramente aquellos pacientes quienes, con toda probabilidad, tenían TIA 1 de aquellos quienes tenían TIA 2. Por lo tanto, grupos heterogéneos de pacientes fueron tratados con una variedad de terapias, haciendo difícil comprender cuál de los tratamientos fueron los más efectivos para cada subtipo.
TIA 1
La descontinuación de AMD, si es posible, es recomendada. Sin embargo, un estudio retrospectivo fue incapaz de documentar una diferencia en el resultado de pacientes en quienes la droga fue suspendida versus aquellos en quienes fue mantenida. Si la droga fue prescrita por una arritmia ventricular que amenazaba la vida, el beneficio de su des-continuación puede no ser mayor como el riesgo de arritmias recurrentes (23,24). Además, debido a sus efectos inhibitorios sobre la conversión de T4 a T3 así como sus efectos beta-bloqueantes, puede ser beneficiosa clínicamente.
Finalmente, aún si la droga es descontinuada, esta se mantiene en la circulación por semanas y aún meses, debido a su almacenamiento en el tejido adiposo. Trip y col comunicaron el tratamiento exitoso de TIA con DAT en dos pacientes, mientras continuaron con AMD (25).
Drogas antitiroideas
Tradicionalmente, grandes dosis de DAT han sido usadas para tratar TIA tipo 1. Tiamazol 40 a 80 mg/día o PTU 400 a 800 mg/día. Mayores dosis de DAT que las usuales son requeridas, debido a que los altos depósitos de yodo intratiroideos antagonizan los efectos inhibitorios de DAT sobre la utilización de yodo tiroideo. Debe prestarse atención sobre la frecuencia de RAM de DAT, incluyendo agranulocitosis ya que son dosis relacionadas. En pacientes quienes fallan para responder a DAT después de 2 a 3 meses de tratamiento, el perclorato de potasio (KClO4) ha sido un adyuvante útil (19,20).
Perclorato de potasio
Es un inhibidor competitivo de la captación tiroidea de I; así, reduce los depósitos de I intratiroideos. La dosis va desde 200 a 1000 mg/día, por algunas semanas a meses; se ha reportado de ser efectivo en normalizar la función tiroidea (26). Perclorato de potasio (KClO4) ha sido asociado con anemia aplásica, pero ningún caso ha sido informado cuando se ha usado para tratar TIA 1. En los pacientes que reciben grandes dosis de DAT más KClO4, parece razonable recomendar el monitoreo bisemanal del recuento leucocitario (27,28).
Yodo radioactivo
Desafortunadamente, I-131 puede raramente ser administrada en la TIA 1, por la baja captación de I debido a los altos niveles de I circulantes provenientes de AMD. En Europa, donde la captación a las 24 horas en TIA 1 es mayor, algunos autores han descrito su uso, pero no es útil en los EE UU (28,29). Una comunicación francesa describió el uso de I-131 en pacientes eutiroideos quienes se habían recuperado de TIA y requerían retomar AMD por recurrencia de arritmias.
La tasa de TIA recurrente en individuos quienes previamente tuvieron un episodio no es claro; aunque algunos han encontrado una tasa de 10%. Este tratamiento ablativo puede ser razonable, pero se dispone de pocos datos para mostrar que la TIA recurrente es un problema clínico (30).
TIA 2
Para pacientes con TIA tipo 2, la prednisona (PDN) es considerada tratamiento de elección, aunque no hay estudios prospectivos controlados con placebo. Algunos pacientes con TIA 2 leve y niveles normales o mínimamente elevados de T4 libre y T3 pueden no requerir tratamiento específico y frecuentemente tienen resolución espontánea de la tiroidi tis. En contraste a lo recomendado en TIA 1, la descontinuación de la droga no es de elección.
Evidencias anecdóticas y publicadas han mostrado que los pacientes frecuentemente mejoran con la terapia corticoide, aunque la terapia con AMD era continuada. Con PDN 40 a 60 mg/día, hay una rápida mejoría en la función tiroidea en la mayoría de pacientes, frecuentemente dentro de una semana (31). La terapia con corticoides debe ser mantenida en niveles relativamente altos por 1 a 2 meses, debido a que pueden ocurrir exacerbaciones de HT si la droga es repuesta rápidamente. Un estudio controlado aleatorizado reciente comparó corticoides (PDN) versus un agente colecistográfico oral (ácido iopanoico), el cual tiene efectos en la conversión de T4 a T3; en esta comunicación, PDN fue más efectiva (tabla 6) (28,32).
Consideraciones terapéuticas
Parece haber casos de TIA mixtas que no responden a la monoterapia, ya sea con DAT o PDN, pero pueden responder a ambos agentes juntos. Estos casos son usualmente identificados después de fallar alguno de estos agentes luego de algunos meses de uso.
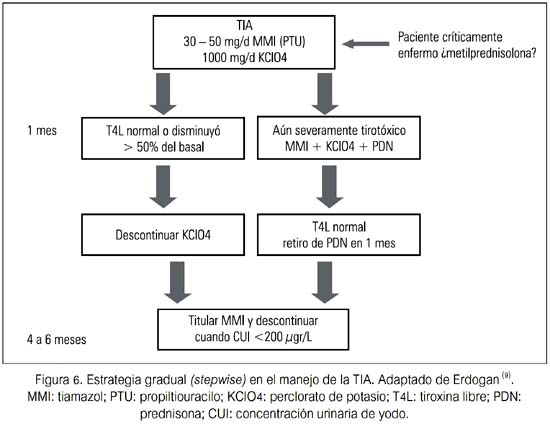
Una estrategia es la gradual (stepwise), donde se agrega una segunda droga después de un periodo de meses durante el cual la función tiroidea no hubiera mejorado. Otra estrategia que algunos recomiendan involucra iniciar en todos los pacientes tanto metimazol 40 mg/d como PDN 40 mg/d. Si hay una respuesta muy rápida (esto es, dentro de 1 a 2 semanas), el paciente probablemente tuvo TIA tipo 2; si no, ambas drogas deberían ser continuadas hasta que mejore la función tiroidea en los siguientes 1 a 2 meses (33,34).
En algunos casos persistentes, donde DAT, PDN ni ambos tienen un impacto significativo sobre la función tiroidea, e incluso si AMD ha sido descontinuada, se ha recomendado otras terapias, como litio, plasmaféresis y tiroidectomía (35,36)
La tiroidectomía está generalmente reservada para pacientes con hipertiroidismo severo, el cual se considera que amenaza la vida y cuando la descontinuación de AMD no es considerada posible desde un punto de vista clínico. Sin embargo, la cirugía en pacientes con hipertiroidismo severo conlleva el riesgo de arritmias y tormenta tiroidea (37-40)
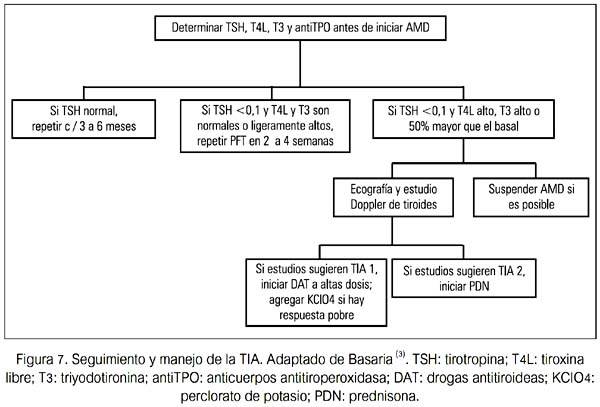
El ácido iopanoico ha sido usado satisfactoriamente para controlar la tirotoxicosis antes de la cirugía. Algunas comunicaciones han descrito tiroidectomía bajo anestesia local, la cual claramente tiene un menor nivel de invasividad y probablemente menor morbilidad(41-44). El seguimiento y manejo de la TIA se presenta en la figura 7.
AMD EN EL EMBARAZO Y LALACTANCIA
El tratamiento de mujeres embarazadas con hipertiroidismo severo ha resultado en hipotiroidismo fetal y bocio obstructivo. Recientemente, Bartalena revisó comunicaciones de 64 neonatos expuestos a AMD in útero, encontrando bocio en solo 3% e hipotiroidismo transitorio en 17% de los niños; leve retardo mental y alteración del habla y del lenguaje fueron encontrados en algunos niños expuestos a AMD in útero (45,46).
Es así que, AMD puede ser razonablemente administrada a mujeres embarazadas si ellas tienen arritmias refractarias y/o que amenazan la vida, y que son resistentes a otros antiarrítmicos.
Es recomendable realizar ecografías obstétricas seriadas para detectar bocio fetal; si un bocio es visto, se debería considerar terapia con LT4 intraamniótica, debido a que esta estrategia ha demostrado ser efectiva (47). En cuanto a la lactancia materna (LM), se sabe que cantidades significativas de AMD y DEA son secretadas en la leche materna. Algunos informes no han mostrado RAM, mientras uno notó hipotiroidismo en un lactante en el cual la LM fue descontinuada y la función tiroidea restaurada. Así, si una madre que toma AMD decide alimentar a su bebé con LM, una evaluación seriada de PFT debe ser realizada en el lactante y el tratamiento apropiado debe iniciarse prontamente si desarrolla hipotiroidismo (48,49)
TAMIZAJE PARA DTIA
Actualmente no existen guías en relación al tamizaje para DTIA. Se recomienda, antes de iniciar la terapia con AMD, realizar un examen tiroideo completo (TSH, T4 libre, T3, y AcT-PO). Esta evaluación basal no solo detectará una disfunción tiroidea subyacente sino también ayuda a identificar a aquellos sujetos quienes pueden estar predispuestos a desarrollar disfunción tiroidea mientras esté con AMD (sujetos con AcTPO +).
A menos que la evaluación clínica lo indique, se debería repetir TSH, T4 L y T3 solo después de tres meses de terapia. En este momento, los cambios esperados en las PFT deberían haber ocurrido; así, estos resultados deben ser considerados basales con AMD. Si las PFT son normales, se recomienda evaluar TSH y T4L cada 3 a 6 meses.
Se debería aconsejar a los pacientes en relación a los síntomas de hipertiroidismo e hipotiroidismo. Una vez que se detecta una DTIA, los pacientes deberían ser referidos a un endocrinólogo para su evaluación.
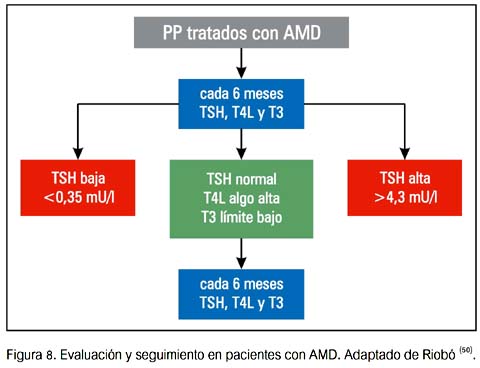
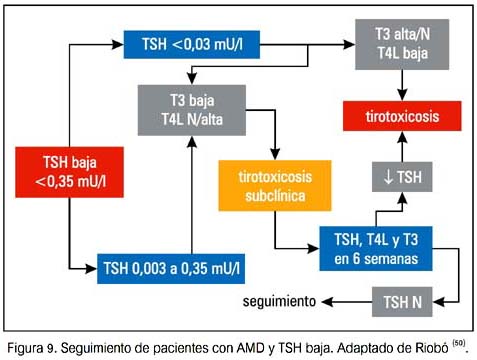
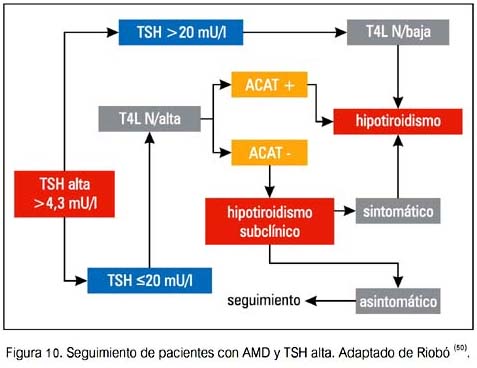
A continuación se presentan algunas sugerencias de seguimiento y manejo de DTAI (50).
REFERENCIAS BIBLIOGRÁFICAS
1. Brunton L, Lazo J, Parker K. Antiarrhythmics. En: Goodman & Gilman. The pharmacological Basis of Therapeutics. 11a edición. 2005.
2. Neal M. Antiarrhythmics drugs. En: Medical pharmacology at a Glance. 4ta edición. Blackwell Science. 2002.
3. Basaria S, Cooper D. Amiodarone and the thyroid. Am J Med. 2005;118:706-14.
4. Martino E, Bartalena L, Braverman L. The effects of amiodarone on the thyroid. Endocrin Rev. 2001;22:240-54.
5. Figge H, Figge J. The effects of amiodarone on thyroid hormone function: a review of physiologyand clinical manifestations. J Clin Pharmacol. 1990;30:588-95.
6. Podrid pJ. Amiodarone: re-evaluation of an old drug.Ann Int Med. 1995;122:689-700.
7. Rajeswaran Ch, Shelton r, Gilbery S. Management of amiodarone-induced thyrotoxicosis. Swiss Med Wkly. 2003;133:579-85.
8. Meurisse M, Gollogly L. Iatrogenic thyrotoxicosis: causal circumstances, pathophysiology, and principles of treatment. review of the literature. World Surg. 2000;24:1377-85.
9. Erdogan M, Gülec S, Gürbuz E. A stepwise approach to the treatment of amiodarone-induced thyrotoxicosis. Thyroid. 2003:13(2):205-9.
10. Ursella S, Testa A,Mazzone M, Silveri G. Amiodarone-induced thyroid dysfunction in clinical practice. Eur revMed pharmacol Sci. 2006;10(5):269-78.
11. Trip MD, Wiersinga W, plomp TA.
Incidence, predictability, and pathogenesis of amiodarone-induced thyrotoxicosis and hypothyroidism. Am J Med.1991;91:507–511.12. Mazonson pD, Williams ML, Cantley LK, Dalldorf FG, Utiger RD, Foster JR. Myxedema coma during long-term amiodarone therapy. Am J Med.1984;77:751–4.
13. Harjai KJ, Licata AA. Effects of amiodarone onthyroid function. Ann Intern Med. 1997;126:63–73.
14. Albert SG. Thyroid dysfunction during chronic AMD therapy. J Am Coll Cardiol. 1987;9:175.
15. Figge J, Dluhy rG. AMD-induced elevation of TSH in patients receiving levothyroxine for primary hypothyroidism. Ann Intern Med. 1990;113:553–5.
16. Martino E, Aghini-Lombardi F, Mariotti S, Bartalena L, Lenziardi M, Ceccarelli C, Bambini G, Safran M, Braverman LE, Pinchera A. Amiodarone iodine-induced hypothyroidism: risk factors and follow-up in 28 cases. Clin Endocrinol(oxf).1987;26(2):227–37.
17. Bartalena L, Grasso, L, Brogioni S, Aghini-Lombardi F, Braverman LE, Martino E. Serum interleukin-6 in amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 1994;78(2):423–7.
18. Pitsiavas V, Smerdely p, Li M, Boyages SC. Amiodarone induces a different pattern of ultrastructural change in the thyroid to iodine excess alone in both the BB/W rat and the Wistar rat. Eur J Endocrinol. 1997;137(1): 89–98.
19. Wiersinga WM. Towards an animal model of amiodarone-induced thyroid dysfunction. Eur J Endocrinol. 1997;137:15–7.
20. Brennan MD, Erickson DZ, Carney JA, Bahn rS.Nongoitrous (type I) amiodarone-associated thyrotoxicosis: evidence of follicular disruption in vitro and in vivo. Thyroid. 1995;5:177–83.
21. Eaton SE, Euinton HA, newman CM, Weetman Ap, Bennet WM. Clinical experience of amiodaroneinduced thyrotoxicosis over a 3-year period: role of colour- flow Doppler sonography. Clin Endocrinol (oxf). 2002;56:33–8.
22. Daniels GH. Amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 2001;86:3–8.
23. Osman F, Franklyn JA, Sheppard MC, Gammage MD. Successful treatment of amiodarone-induced thyrotoxicosis. Circulation. 2002;105:1275–7.
24. Moolman J. Thyroid hormone and the heart. Cardiovasc J South Africa. 2002;13:159-63.
25. Trip MD, Duren Dr, Wiersinga WM. Two cases of amiodarone induced thyrotoxicosis successfully treated with a short course of antithyroid drugs while amiodarone was continued. Br Heart J. 1994;72:266–8.
26. Reichert LJ, de rooy HA.
Treatment of amiodaroneinduced hyperthyroidism with potassium perchlorateand methimazole during amiodarone treatment.BMJ. 1989;298:1547-8.27. Newnham HH, Topliss DJ, Le Grand BA, Chosich N, Harper RW, Stockigt JR. Amiodarone-induced hyperthyroidism: assessment of the predictive valueof biochemical testing and response to combined therapy using propylthiouracil and potassium perchlorate. Aust n Z J Med. 1988;18:37–44.
28. Bartalena L, Brogioni S, Grasso L, Bogazzi F,Burelli A, Martino E. Treatment of amiodaroneinduced thyrotoxicosis, a difficult challenge: results of a prospective study. J Clin Endocrinol Metab.1996;81(8):2930–3.
29. Hermida JS, Jarry G, Tcheng E, Moullart V, Arlot S, Rey JL, Delonca J, Schvartz C. Radioiodine ablation of the thyroid to allow the reintroduction of amiodarone treatment in patients with a prior history of amiodarone-induced thyrotoxicosis. Am J Med. 2004;116(5)16:345–8.
30. Ryan LE, Braverman LE, Cooper DS, LadensonpW, Kloos rT.
Can amiodarone be restarted after amiodarone-induced thyrotoxicosis? Thyroid. 2004;14:149–53.31. Daniels GH. Amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab.2001;86:3–8.
32. Bogazzi F, Bartalena L, Cosci C, Brogioni S,Dell'unto E, Grasso L, Aghini-Lombardi F, rossi G, pinchera A, Braverman LE, Martino E. Treatmentof type II amiodarone-induced thyrotoxicosis by either iopanoic acid or glucocorticoids: a prospective, randomized study. J Clin Endocrinol Metab. 2003;88:1999–2002.
33. Daniels GH. Amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 2001;86:3–8. 37–42.
34. Broussolle C, Ducottet X, Martin C, Barbier Y, Bornet H, noel G, orgiazzi J. rapid effectiveness of prednisone and thionamides combined therapy in severe amiodarone iodine-induced thyrotoxicosis. Comparison of two groups of patients with apparently normal thyroid glands. J Endocrinol Invest. 1989;12(1):37–42.
35. Samaras K, Marel GM. Failure of plasmapheresis, corticosteroids and thionamides to ameliorate a case of protracted amiodarone-induced thyroiditis. Clin Endocrinol (oxf). 1996;45:365–8.
36. Aghini-Lombardi F, Mariotti S, Fosella pV, GrassoL, pinchera A, Braverman LE, Martino E. Treatment of amiodarone iodine-induced thyrotoxicosis with plasmapheresis and methimazole. J Endocrinol Invest. 1993;16(10):823–6.
37. Brennan MD, van Heerden JA, Carney JA.Amiodarone-associated thyrotoxicosis (AAT):experience with surgical management. Surgery.1987;102:1062–7.
38. Mulligan DC, McHenry Cr, Kinney W, EsselstynCB Jr. Amiodarone-induced thyrotoxicosis: clinical presentation and expanded indications for thyroidectomy. Surgery. 1993;114(6):1114–9.
39. Hamoir E, Meurisse M, Defechereux T, Joris J,Vivario J, Hennen G. Surgical management of amiodarone-associated thyrotoxicosis: too risky or too effective? World J Surg. 1998;22(6):537–542.
40. Meurisse M, Hamoir E, DSilva M, Joris J, Hennen G. Amiodarone-induced thyrotoxicosis: is there a place for surgery? World J Surg. 1993;17:622–6.
41. Bogazzi F, Aghini-Lombardi F, Cosci C, Lupi I, Santini F, Tanda ML, Miccoli p, Basolo F, pinchera A, Bartalena L, Braverman LE, Martino E. Iopanoic acid rapidly controls type I amiodarone-induced thyrotoxicosis prior to thyroidectomy. J Endocrinol Invest. 2002;25(2):176–80.
42. Bogazzi F, Miccoli p, Berti p, Cosci C, Brogioni S, Aghini-Lombardi F, Materazzi G, Bartalena L, pincheraA, Braverman LE, Martino E. preparation with iopanoic acid rapidly controls thyrotoxicosis in patients with amiodarone-induced thyrotoxicosis beforethyroidectomy. Surgery. 2002;132(6):1114 –7.
43. Klein SM, Greengrass rA, Knudsen n, Leight G, Warner DS. Regional anesthesia for thyroidectomy in two patients with amiodarone-induced hyperthyroidism. Anesth Analg. 1997;85(1):222–224.
44. Mehra A, Widerhorn J, Lopresti J, rahimtoola SH. Amiodarone-induced hyperthyroidism: thyroidectomy under local anesthesia. Am Heart J. 1991;122(4 pt 1):1160–1.
45. Vicens-Calvet E, potau n, Carreras E, Bellart J,Albisu MA, Carrascosa A. Diagnosis and treatment in utero of goiter with hypothyroidism caused byiodide overload. J pediatr. 1998;133(1):147–8.
46. Bartalena L, Bogazzi F, Braverman LE, Martino E. Effects of amiodarone administration during pregnancy on neonatal thyroid function and subsequent neurodevelopment. J Endocrinol Invest. 2001;24(2):116–30.
47. De Catte L, De Wolf D, Smitz J, Bougatef A, De Schepper J, Foulon W. Fetal hypothyroidism as acomplication of amiodarone treatment for persistentfetal supraventricular tachycardia. Prenat Diagn. 1994;14(8):762–5.
48. McKenna WJ, Harris L, rowland E, Whitelaw A, Storey G, Holt D. Amiodarone therapy during pregnancy. Am J Cardiol. 1983;51(7):1231–1233.
49. Hall CM, McCormick KpB. Amiodarone and breast feeding. Arch Dis Child Fetal Neonatal Ed. 2003;88:F255-4.
50. riobó p. Amiodarona y tiroides. Simposium Fibrilación auricular: Nuevos horizontes. Fundación Jiménez Díaz. 2003.
Trabajo recibido el 31 de agosto de 2010 y aceptado para publicación el 29 de setiembre de 2010.
Correo electrónico: jlpi_09@hotmail.com

