










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. vol.77 no.1 Lima ene./mar. 2016
http://dx.doi.org/10.15381/anales.v77i1.11555
10.15381/anales.v77i1.11555
CASOS CLÍNICOS
Insulinoma pancreático gigante
Giant pancreatic insulinoma
Fernando Revoredo Rego1, José De Vinatea De Cárdenas1, Gustavo Reaño Paredes1, Luis Villanueva Alegre1, Fritz Kometter Barrios1, Jorge Tang Sing1, Mónica Uribe León1, José Arenas Gamio2
1 Servicio de Cirugía de Páncreas, Bazo y Retroperitoneo, Hospital Nacional Guillermo Almenara Irigoyen, EsSalud, Lima, Perú.
2 Servicio de Anatomía Patológica, Hospital Nacional Guillermo Almenara Irigoyen, EsSalud, Lima, Perú.
Resumen
Los insulinomas son los tumores neuroendocrinos pancreáticos más frecuentes y típicamente son benignos y pequeños. Los insulinomas gigantes de páncreas son tumores raros. Reportamos el caso de un paciente varón de 67 años, quien presentaba síntomas y signos de hipoglicemia. Subsecuentes estudios de laboratorio y radiológicos establecieron el diagnóstico de un insulinoma de 13,5 cm. Fue sometido a pancreaticoduodenectomía. A pesar del tamaño, en la evaluación patológica no se identificó invasión local ni metástasis. Además de la descripción de la presentación clínica y los hallazgos operatorios, se realiza una revisión de las opciones para la localización y manejo de los insulinomas.
Palabras clave: Insulinoma, Gigante; Páncreas, Neoplasias; Tumores Neuroendocrinos.
Abstract
Insulinomas are the most common pancreatic neuroendocrine tumors and are typically small and benign. Giant pancreatic insulinomas are rare pancreatic tumors. We report a 67 year old man who presented with signs and symptoms of hypoglycemia. Subsequent laboratory and radiologic studies established the diagnosis of a 13.5 cm insulinoma. The patient underwent a pancreatoduodenectomy. Despite the size, neither local invasion nor metastatic disease was identified on pathological evaluation. In addition to describing the clinical presentation and operative findings, localization and management options of insulinomas are reviewed.
Keywords: Insulinoma, Giant; Pancreatic Neoplasms; Neuroendocrine Tumors.
INTRODUCCIÓN
Los tumores neuroendocrinos son neoplasias caracterizadas por su capacidad de almacenar y secretar diferentes péptidos y neuroaminas. Algunas de estas sustancias causan síndromes clínicos específicos mientras que otras están elevadas en el plasma o la orina y no se asocian a síndromes o síntomas específicos (1).
Los tumores neuroendocrinos de páncreas (TNEP) tienen una incidencia estimada en 1 por 100 000 habitantes (1,2) y se dividen en dos grupos: aquellos asociados con un síndrome funcional debido a la secreción ectópica de una sustancia activa biológica (tumores funcionantes) y aquellos que no están asociados con un síndrome funcional (usualmente llamados no funcionantes) (1,2). Los TNEP funcionantes incluyen a los insulinomas, gastrinomas, VIPomas, somatostatinomas, glucagonomas, TNEP que causan liberación de la hormona de crecimiento y un grupo menos común de TNEP que secretan ACTH (ACTHomas) y causan síndrome de Cushing, TNEP que causan síndrome carcinoide, TNEP que causan hipercalcemia y muy raramente TNEP ectópicos que secretan hormona luteinizante, renina o eritropoyetina. Los TNEP (funcionantes y no funcionantes) secretan también otras sustancias (cromogranina, sinaptofisina, enolasa neuronal específica, subunidades de la gonadotropina coriónica humana, neurotensina, grhelina), sin embargo, estas no causan un síndrome hormonal especifico (2,3). Los insulinomas son los TNEP funcionantes más frecuentes, con una incidencia reportada de 4 casos por millón de habitantes por año, con un pico de incidencia específica en la quinta década de la vida y con una leve predilección por el sexo femenino (4).
Más del 90% de los insulinomas son tumores pequeños y únicos; aproximadamente 10% son múltiples, menos del 10% son malignos (clásicamente determinada por la evidencia de invasión local o metástasis) y del 5% al 10% están asociados con la neoplasia endocrina múltiple tipo 1 (NEM – 1) (4-6) o síndrome de Wermer, desorden autosómico dominante asociado con mutaciones del gen NEM – 1 (brazo largo del cromosoma 11q13), caracterizada por hiperplasia tiroidea, adenomas de la pituitaria anterior y tumores del páncreas endocrino y duodeno (7). Los insulino mas son múltiples, con mayor frecuencia en pacientes con NEM – 1 (5).
El 43% de insulinomas está localizado en la cabeza y cuello del páncreas, el 30,9% en la cola y 25,3% en el cuerpo. Se emplazan en diferentes partes del páncreas debido a la igual distribución de las células β en el parénquima pancreático. Menos del 1% son ectópicos (extrapancreáticos). Los insulinomas ectópicos con síntomas de hipoglicemia son raros y más comúnmente se encuentran en la pared duodenal, íleon, yeyuno, pared gástrica, hilio esplénico, ligamento gastroesplénico, pulmón, cérvix y ovario (7).
Los pacientes con insulinomas característicamente presentan síntomas de hipoglicemia, especialmente síntomas neuroglicopénicos (confusión, alteración de conciencia, ansiedad, mareos, cambios en la personalidad, incoherencia, visión borrosa, convulsiones y coma), así como signos y síntomas simpático adrenales (palpitaciones, debilidad, sudoración, temblores y taquicardia), como consecuencia de la liberación de catecolaminas en respuesta a la hipoglicemia (1,4,5), los cuales usualmente empeoran con el ayuno (1). La duración de los síntomas antes del diagnóstico varía desde muchos meses a muchas décadas (7).
En 1935, Whipple y Frantz describen la triada de hallazgos clínicos en pacientes con insulinomas: síntomas de hipoglicemia, niveles plasmáticos de glucosa bajos y que se resuelve con la ingesta de carbohidratos (8), característica distintiva de los insulinomas y que se conoce como la ‘triada de Whipple’. Actualmente existen 6 criterios para el diagnóstico de insulinoma: niveles de glucosa sanguínea de 2,2 mmol/L o menos (≤ 40 mg/dL). Concomitantemente, niveles de insulina de 6 μU/ mL o mayores (≥ 36 pmol/L), niveles de péptido C de 200 pmol/L o mayores, niveles de proinsulina de 5 pmol/L o mayores, niveles de β hidroxibutirato de 2,7 mmol/L o menores y ausencia de sulfonilureas (metabolitos) en el plasma o la orina (1,9). Pruebas posteriores controladas bajo supervisión incluyen la prueba de ayuno de 72 horas (10), que es el criterio estándar para establecer el diagnóstico de insulinoma (1,9). Inicialmente se debe medir la glucosa sanguínea, insulina, péptido C y proinsulina cada 6 horas hasta que la glucosa sanguínea sea de 60 mg/dL o menos, tras lo cual las mediciones se deben hacer cada 1 o 2 horas hasta que los niveles de glucosa lleguen a 2,2 mmol/L o menos (≤ 40 mg/dL) y la prueba de ayuno debe ser terminada (10).
Desde el primer reporte de la cura del insulinoma mediante cirugía, realizada por Roscoe Graham en Toronto y publicada por Howland en 1929 (8,11), se considera que el único tratamiento curativo para el insulinoma es la cirugía. El manejo médico es usualmente considerado en pacientes en quienes no se encontraron los insulinomas durante la cirugía y en aquellos que se niegan a la cirugía o no son candidatos para la misma (7).
La gran mayoría de los insulinomas son muy pequeños, siendo en 83,6% iguales o menores a 2 cm de diámetro, en 8,2% de 2,1 cm a 3 cm de diámetro y en 8,2% mayores a 3 cm de diámetro (7); sin embargo, también hay reportes de insulinomas de hasta 21 cm de diámetro (4). Estos tumores inusualmente grandes son extremadamente raros y aunque no se precisa exactamente el tamaño para definirlos, algunos autores definen como ‘insulinomas gigantes’ a aquellos mayores de 9 cm en su diámetro mayor. En las series de insulinomas desde 1927 hasta la fecha se ha reportado solamente 39 casos de insulinomas gigantes (4,12-14). Comunicamos un nuevo caso de insulinoma gigante de 13,5 cm de diámetro mayor, sin evidencia de invasión local o metástasis.
REPORTE DE CASO
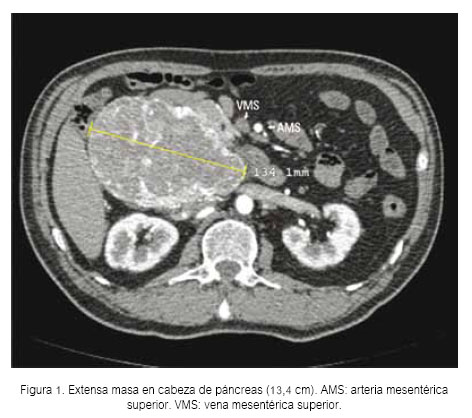
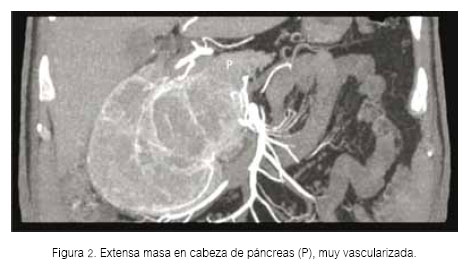
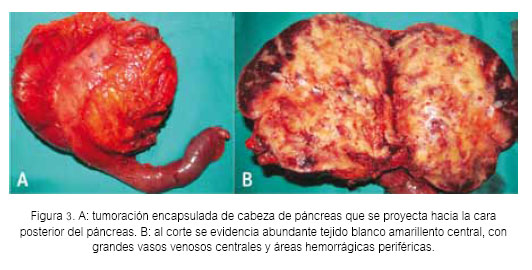
Paciente varón de 67 años, casado, con grado de instrucción superior, quien refiere que 9 meses antes de su ingreso al Hospital Nacional Guillermo Almenara Irigoyen (HNGAI), presentaba episodios de sudoración por las mañanas, que se resolvían con la ingesta del desayuno. Además, durante el medio día tenía visión borrosa que se solucionaba con la ingesta del almuerzo. Siete meses antes de su ingreso, se asoció sensación de intranquilidad, por lo que acudió al consultorio de Psiquiatría de otro hospital donde le diagnosticaron depresión y le dieron tratamiento sertralina, alprazolam y amisulpiride. La sintomatología descrita se fue haciendo más frecuente y dos meses antes del ingreso acudió a la Emergencia de otro hospital debido a que presentaba visión borrosa, debilidad generalizada y sudoración. En dicha oportunidad le realizaron un glucotest y se registró una glicemia de 43 mg/dL. Se le colocó dextrosa endovenosa, con lo que se resolvieron los síntomas y el paciente fue dado de alta. Un mes antes de su ingreso, los síntomas se presentaban todos los días, por lo que fue hospitalizado para estudio y posteriormente referido al HNGAI para manejo. A su ingreso al servicio de Endocrinología del HNGAI, las funciones vitales eran estables (frecuencia cardíaca 74/minuto, frecuencia respiratoria 18/minuto y la presión arterial 110/60 mmHg). Lo único relevante en el examen físico fue el hallazgo de una masa móvil de 10x8 cm en mesogastrio, no dolorosa a la palpación. Se realizaron los siguientes exámenes de laboratorio de rutina: leucocitos (13 700), hemoglobina (15,5 g/dL), glucosa (85 mg/dL), urea (15,41 mg/dL), creatinina (0,8 mg/dL), sodio (140,7 mmol/L), potasio (4,52 mmol/L), cloro (107 mmol/L), fosfatasa alcalina (77 U/L), gammaglutamiltranspeptidasa (72 U/L), bilirrubina total (0,77 mg/dL), albúmina (4,3 g/dL), INR (0,97), CA 19,9 (2 U/mL), AFP (3,05 ng/mL), CEA (1,9 ng/mL). En la tomografía abdominal (Figuras 1 y 2) se reportó una masa en cabeza de páncreas de 12,6 x 13,5 x 9,4 cm, de contornos regulares y definidos. No condicionaba dilatación de las vías biliares ni del conducto de Wirsung. Con estos exámenes se hicieron los siguientes diagnósticos: 1) Hiperinsulinismo endógeno, 2) Masa en mesogastrio a descartar un insulinoma y 3) Trastorno depresivo. Se solicitó prueba de ayuno, cuyos resultados fueron los siguientes: glucosa en 31 mg/dL, insulina 156 μU/ mL y péptido C 13,4 ng/mL. Con estos resultados se hizo el diagnóstico de insulinoma y se trasfirió al Servicio de Cirugía de Páncreas para manejo, donde luego de completar los exámenes pre operatorios se programó para cirugía. Se realizó una pancreaticoduodenectomía con resección pilórica y reconstrucción en un asa. Los hallazgos descritos (Figura 3) correspondieron a una tumoración de superficie multilobulada, encapsulada, neovascularizada de 13,5 cm de diámetro. Al corte era completamente sólida, con abundante tejido blanco amarillento central y grandes vasos venosos centrales y áreas hemorrágicas periféricas. El tumor se originaba en la cabeza del páncreas, proyectándose hacia la cara posterior y hacia el proceso uncinado. Se asentaba por delante de la vena cava inferior y se adhería a la cara lateral derecha y posterior, comprimiendo y elongando el tronco venoso porto – mesentérico. No se apreció metástasis hepáticas. El paciente estuvo hospitalizado en la unidad de cuidados intensivos los primeros dos días del postoperatorio y posteriormente pasó a piso, en donde luego de una evolución favorable fue dado de alta en día 12 del postoperatorio.
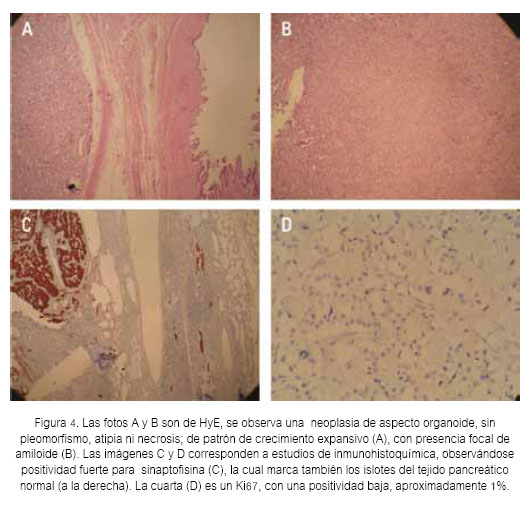
El reporte de anátomo patología (Figura 4) correspondió a una neoplasia endocrina pancreática bien diferenciada compatible con insulinoma. La inmunohistoquímica fue: cromogranina (+), sinaptofisina (+), Ki67 < 1%, y una mitosis por 10 campos de alto poder. No hubo invasión vascular ni perineural; y ganglios peripancreáticos y regionales sin evidencia de malignidad (0/7).
DISCUSIÓN
Los insulinomas pancreáticos son neoplasias raras cuya sintomatología básicamente está relacionada con la hipoglicemia. En el caso que presentamos hubo síntomas neuroglicopénicos (visión borrosa, cambios en la personalidad), así como síntomas simpáticos adrenales (sudoración, debilidad). Se hizo el diagnóstico de depresión y se le dio tratamiento farmacológico. Muchas veces los pacientes tienen una sintomatología que se confunde con problemas psiquiátricos y pueden pasar muchos meses o incluso décadas hasta que se hace el diagnóstico definitivo (7).
El mecanismo por el cual los insulinomas mantienen altos niveles de secreción de insulina en presencia de hipoglicemia es desconocido aún (12). Por otro lado se ha descrito que el amiloide está presente en más de 50% de los insulinomas. El componente más importante del amiloide es un polipéptido de 37 aminoácidos conocido como polipéptido amiloide de los islotes o amilina (5). La sobreproducción, el aberrante procesamiento y secreción del amiloide por las células β anormales del insulinoma, han sido postulados como la causa de la amiloidogénesis (5). Con respecto a los insulinomas gigantes, más del 70% contienen a este elemento (5,12).
A nuestro paciente se le realizó la prueba de ayuno, la que salió positiva; y con los hallazgos en la tomografía fue sometido a una pancreaticoduodenectomía. La resección quirúrgica es la única forma de cura. Para ello, es necesario localizar preoperatoriamente la lesión de manera de planificar un abordaje quirúrgico apropiado, tratando siempre de preservar la mayor parte del tejido pancreático sano (7).
La localización preoperatoria se puede dividir en dos tipos de métodos: no invasivos o invasivos. Los métodos no invasivos incluyen ultrasonografía abdominal, tomografía computarizada y resonancia magnética. Los métodos invasivos incluyen la angiografía selectiva, ultrasonografía endoscópica, toma de muestras venosas portales transhepáticas, estimulación arterial selectiva con calcio y la cintigrafía de receptores de somatostatina (2-4,7). En una revisión sistemática de más de 6 000 insulinomas, Mehrabi encontró una sensibilidad de 32,6% para la ultrasonografía, 43,9% para la tomografía, 53,3% para la resonancia magnética, 58,5% para la angiografía selectiva, 74,8% para la ultrasonografía endoscópica, 77,9% para la toma de muestras venosas portales transhepáticas, 89,2% para la estimulación arterial selectiva con calcio y 19,5% para la cintigrafía de receptores de somatostatina (7). Actualmente se acepta a la tomografía como el paso inicial estándar en la detección de los insulinomas; además, permitirá determinar la multiplicidad, las metástasis o la proximidad a vasos mayores o al conducto pancreático principal (7).
La localización intraoperatoria incluye la inspección, palpación, ultrasonografía intraoperatoria y monitoreo de la glucosa en el intraoperatorio. Mehrabi encontró una sensibilidad de 81,4% para la inspección y palpación, 91,2% para la ecografía intraoperatoria y 76,9% para el monitoreo de la glucosa en el intraoperatorio. La ecografía intraoperatoria tiene especial importancia en determinar la proximidad de la lesión con la vía biliar o el conducto pancreático principal, sobre todo si se va a realizar una enucleación (7). Se ha descrito que 10% a 27% de los insulinomas permanecen indetectables en el intraoperatorio (inspección y palpación), si no se tiene una localización pre operatoria (7).
En el abordaje quirúrgico abierto, el páncreas completo debe ser inspeccionado y palpado cuidadosamente. Luego se debe realizar una ultrasonografía en el hígado y el páncreas para excluir enfermedad metastásica o tumores múltiples, así como para determinar la distancia entre el margen del tumor y el conducto pancreático principal. Una vez que se encuentra el tumor, y si la enucleación es factible, se la debe realizar. La enucleación es el procedimiento de elección, especialmente para tumores pequeños (< 2 cm), benignos, solitarios y superficiales (15). Si el tumor está localizado profundamente en el parénquima pancreático y anatómicamente no es factible la enucleación, se debe realizar una resección segmentaria, pancreatectomía distal (preservando bazo) o una duodenopancreatectomía, como alternativas seguras y efectivas. Para los tumores con sospecha de malignidad o tumores grandes que comprometen grandes segmentos del páncreas, se debe realizar una resección formal más que una enucleación. En casos de múltiples lesiones, una pancreatectomía total es la mejor opción. Sin embargo, también se ha reportado la enucleación de las lesiones de la cabeza asociada a una pancreatectomía distal (7,15). En nuestro paciente, debido al tamaño del tumor (13,5 cm) no se tuvo otra alternativa que una resección formal.
Con el desarrollo de la cirugía laparoscópica se reportó la primera cirugía exitosa para un insulinoma pancreático en 1996, por Gagner (16). El abordaje laparoscópico es un procedimiento seguro y factible en pacientes con tumores pequeños, benignos y solitarios localizados en el cuerpo o la cola del páncreas. Este procedimiento tiene la ventaja de acortar la estancia hospitalaria y un período de recuperación más rápido (17). Liu (17), en una revisión de 52 casos, no encontró diferencias entre las enucleaciones laparoscópicas o abiertas en cuanto a tiempo operatorio, sangrado y morbilidad. Aunque se ha reportado la enucleación laparoscópica de insulinomas localizados en la parte dorsal del páncreas o de localización profunda (18), la mayoría de autores está de acuerdo que el abordaje abierto debe ser el de elección en estos casos (19).
La morbilidad del abordaje quirúrgico abierto y laparoscópico es de 35,4% y 32,8%, respectivamente (7), siendo la fistula pancreática la complicación quirúrgica más importante, la cual ocurre con mayor frecuencia después de una enucleación; sin embargo, tienen un mejor pronóstico que las fistulas asociadas a otras resecciones (15).
La mortalidad posquirúrgica reportada es 3,7% para la cirugía abierta mayormente asociada con insulinomas malignos y metastásicos y 0% para el abordaje laparoscópico (7).
Se han postulado tres hipótesis sobre el origen de los insulinomas gigantes. En la primera, es posible que un insulinoma con pobre producción de insulina crezca muy lentamente durante un largo período de tiempo sin causar síntomas, hasta que alcanzan un gran tamaño capaz de producir suficiente insulina para inducir los ataques de hipoglicemia (12,20). En la segunda, es posible que haya un cambio fenotípico de un TNEP no funcionante que facilite su transformación a un tumor productor de insulina (21). Y la tercera es debido a la sobreproducción de amiloide. Con respecto a esta última hipótesis hay algunos reportes de insulinomas gigantes sin depósitos de amiloide significativos (4).
El porcentaje de malignidad en los insulinomas es de menos del 10%. No hay marcadores definitivos para distinguir entre los insulinomas benignos y los malignos. Sin embargo, los tumores malignos están asociados mayormente a metástasis en el hígado y en los ganglios linfáticos (37,8% y 18,4%, respectivamente). La enfermedad metastásica usualmente empeora la situación del paciente, aún después de la resección, y el pronóstico es pobre (7).
Actualmente, la Organización Mundial de la Salud (OMS) ha clasificado a los TNEP usando un sistema híbrido que incorpora información del estadiaje (TNM) y grados (de acuerdo al índice mitótico y el Ki67). Estos sistemas han permitido una estratificación pronóstica (22). Así tenemos que la sobrevida a 5 años, para un estadio I es de 61%, para un estadio II es de 52%, para un estadio III es de 41% y para un estadio IV es de 15% (23). Cabe resaltar que la clasificación TNM de la OMS es distinta a la de la Sociedad Europea de Tumores Neuroendocrinos (ENETS).
De acuerdo a la clasificación TNM de la OMS, nuestro paciente sería un T3 (el tumor se extiende más allá del páncreas, pero sin comprometer el tronco celíaco o la arteria mesentérica superior), N0 (sin ganglios comprometidos), M0 (sin metástasis). Estadio IIA.
En cuanto a la clasificación por grados (G1: bajo grado, < 2 mitosis/10 campos de alto poder y/o Ki67 < 2%; G2: grado intermedio, 2 a 20 mitosis/10 campos de alto poder y/o Ki67 3% a 20%; G3: carcinoma neuroendocrino, alto grado, > 20 mitosis/10 campos de alto poder y/o Ki67 > 20%), se la considera como un predictor fundamental de resultados, que estima la agresividad biológica (potencial de diseminación metastásica) (24). Para páncreas, se ha demostrado que el valor sobre el cual el riesgo de muerte se incrementa estadísticamente significativamente es un Ki 67 de 5%. Este valor debe ser tomado en cuenta específicamente en las decisiones de los pacientes G2 (25). De acuerdo al grado, nuestro paciente sería un G1 (Ki67 < 1%, y 1 mitosis/10 campos de alto poder).
De los 39 insulinomas gigantes reportados, solo 2 tuvieron un Ki67 de 20% (4,12). La prevalencia de enfermedad metastásica fue de 54% (21 de 39) (4,12-14) en el momento del diagnóstico, cifra mucho mayor que el 10% de lo reportado en la mayoría de insulinomas (no gigantes) (4).
Si bien es cierto el abordaje quirúrgico es el único método que cura, el manejo médico debe considerarse en casos de enfermedad metastásica, tumores no resecables o como tratamiento de emergencia en período preoperatorio (7). La terapia más efectiva para controlar la hipoglicemia es el diazóxido. Este suprime la secreción de insulina por acción directa en las células β y aumenta de la glucogenólisis. El diazóxido controla la hipoglicemia en 50% a 60% de los pacientes y ha sido efectivo por más de 20 años en algunos de ellos (2). Los análogos de la somatostatina de larga duración (octreotide, lanreotide) también pueden usarse para controlar la hipoglicemia, con una efectividad de 35% a 50%. Sin embargo, deben utilizarse con cuidado, ya que en algunos pacientes pueden empeorar la hipoglicemia debido a la inhibición de los mecanismos contrarreguladores (2). Los corticoides son otra opción de tratamiento médico, pero su uso es limitado por su efecto limitado y transitorio. La estreptozotocina, antibiótico derivado del hongo Streptomyces achromogenes, inhibe la biosíntesis de insulina en las células β. Se aplica vía endovenosa o por cateterización del tronco celíaco en casos de enfermedad metastásica. También se han mencionado el uso de verapamilo y fenitoína como terapia médica (7). Recientemente, el everolimus, inhibidor del complejo mTORC1 (diana de rapamicina en células de mamífero), una serina-treonina quinasa que tiene un papel central en la regulación del crecimiento, proliferación y supervivencia celular (26), ha demostrado ser efectivo en el control de la hipoglicemia en pacientes con insulinomas malignos o que no pueden ser resecados (26,27). Se piensa también que el sunitinib, un inhibidor tironsinaquinasa, tiene un rol inhibiendo la angiogénesis, a través de la vía del factor de crecimiento endotelial (13). Ha demostrado mejorar la sobrevida libre de enfermedad con una media de 11,4 meses comparado con 5,5 meses en pacientes con placebo (28).
La tasa de cura es de 93% y la tasa de recurrencia de 7,2%. Se recomienda un seguimiento regular de los pacientes después de la cirugía (3, 6 y 12 meses y luego anualmente), especialmente en pacientes con insulinomas malignos y NEM – 1, debido a la alta tasa de recurrencia. Si la recurrencia no aparece en 20 años, la probabilidad de que recurra es extremadamente baja (7).
Para concluir, podemos decir que los insulinomas gigantes del páncreas son tumores extremadamente raros, con un mayor potencial de malignidad. Sin embargo, en algunos casos, a pesar de su gran tamaño, no presentan metástasis ni criterios de agresividad biológica, en el momento del diagnóstico. El tratamiento de elección es la resección quirúrgica con la remisión completa de los síntomas.
REFERENCIAS BIBLIOGRÁFICAS
1. Vinik AI, Woltering EA, Warner RR, Caplin M, O´Dorisio TM, Wiseman GA, et al. NANETS consensus guidelines for the diagnosis of neuroendocrine tumors. Pancreas. 2010;39(6):713–34. DOI: 10.1097/MPA.0b013e3181ebaffd. [ Links ]
2. Metz DC, Jensen RT. Gastrointestinal neuroendocrine tumors: pancreatic endocrine tumors. Gastroenterology. 2008;135:1469 – 1492. DOI: 10.1053/j.gastro.2008.05.047. [ Links ]
3. Falconi M, Plockinger U, Kwekkeboom DJ, Manfredi R, Korner M, Kvols L, et al. Well differentiated pancreatic nonfunctioning tumor/carcinoma. Neuroendocrinology. 2006;84:196–211. [ Links ]
4. Callacondo D, Arenas JL, Ganoza AJ, Rojas – Camayo J, Quezada – Olarte J, Robledo H. Giant insulinoma. A report of 3 cases and review of the literatura. Pancreas. 2013;43(8):1323–32. DOI: 10.1097/MPA.0b013e318292006a. [ Links ]
5. Mittendorf EA, Liu Y, Mc Henry C. Giant insulinoma: case report and review of the literature. J Clin Endocrinol Metab. 2005;90:575–80. [ Links ]
6. Marek B, Kajdaniuk D, Kos-Kudla B, Foltyn W, Borgiel– Marek H, Matyia V, et al. Insulinoma – diagnosis and treatment. Endokrynol Pol. 2007;58:58–62. [ Links ]
7. Mehrabi A, Fischer L, Hafezi M, Dirlewanger A, Grenacher L, Diener MK, et al. A systematic review of localization, surgical treatment options, and outcome of insulinoma. Pancreas. 2014;43:675–86. DOI: 10.1097/MPA.0000000000000110. [ Links ]
8. Whipple AO, Frantz VK. Adenoma of islet cells with hyperinsulinism: a review. Ann Surg. 1935;101:1299–335. [ Links ]
9. De Heeder WW, Niederle B, Scoazee JY, Pauwels S, Kloppel G, Falconi M, et al. Well differentiated pancreatic tumor/carcinoma: insulinoma. Neuroendocrinology. 2006;84:183–8. [ Links ]
10. Service FJ. Hypoglicemic disorders. N Engl J Med. 1995;332:1144–52. [ Links ]
11. Howland G, Campbell WR, Maltby EJ, Robinson WL. Dysinsulinism: convulsions and coma due to islet cell tumor of the pancreas with operation and cure. JAMA. 1929;93:674–9. DOI: 10.1001/ jama.1929.02710090014006. [ Links ]
12. Ielpo B, Caruso R, Ferri V, Quijano Y, Duran H, Diaz E, et al. Giant pancreatic insulinoma. The bigger the worse?. Report of two cases and literarture review. Int J Surg Case Rep. 2013;4(3):265–8. DOI: 10.1016/j.ijscr.2012.11.019. [ Links ]
13. Fenech VA, Ellul P, Abela A, Caruana C, Cassar M, Laferla G. A rare finding of a rare disease. A case report of a giant insulinoma. Pancreas. 2013;42(7):1195–6. DOI: 10.1097/ MPA.0b013e31829004d9. [ Links ]
14. Karavias D, Habeos I, Maroulis I, Kalogeropoulou C, Tsamandas A, Chaveles I, et al. Giant malignant insulinoma. Ann Surg Treat Res. 2015;88(5):289– 93. DOI: 10.4174/astr.2015.88.5.289. [ Links ]
15. Hackert T, Hinz U, Fritz S, Strobel O, Schneider L, Hartwig W, et al. Enucleation in pancreatic surgery: indications, technique, and outcome compared to standard pancreatic resections. Langenbecks Arch Surg. 2011;396:1197–203. DOI: 10.1007/ s00423-011-z. [ Links ]
16. Gagner M, Pomp A, Herrera M. Early experience with laparoscopic resections of islet cell tumors. Surgery. 1996;120:1051–4. [ Links ]
17. Liu H, Peng C, Zhang S, Wu Y, Fang H, Sheng H, et al. Strategy for the surgical management of insulinomas: analysis of 52 cases. Dig Surg. 2007;24:463–70. [ Links ]
18. Isla A, Arbuckle JD, Kekis PB, Lim A, Jackson JE, Todd JF, et al. Laparoscopic management of insulinomas. Br J Surg. 2009;96:185–90. DOI: 10.1002/jbs.6465. [ Links ]
19. Zhao YP, Zhan HX, Zhang TP, Cong L, Dai MH, Liao Q, et al. Surgical management of patients with insulinomas: result of 292 cases in a single institution. J Surg Oncol. 2011;103:169–74. DOI: 10.1002/jso.21773. [ Links ]
20. Sugiyama T, Kouyama R, Tani Y, Izumiyama H, Akashi T, Kishimoto S, et al. Giant malignant insulinoma which developed from a non-functioning pancreatic tumor over a long period of time. Intern Med. 2010;49:1573–9. [ Links ]
21. Vasi PG, Gupta D, Dahlk S. A unique case of nonfuntional metastatic pancreatic neuroendocrine tumor transforming into an insulin secreting tumor with an unusual clinical course. Pancreas. 2011;40:781–4. DOI: 10.1097/MPA.0b013e318212c42d. [ Links ]
22. Klimstra DS, Modlin IR, Coppola D, Lloyd RV, Suster S. The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems. Pancreas. 2010;39(6):707–12. DOI: 10.1097/MPA.0b013e3181ec124e. [ Links ]
23. Bilimoria KY, Bentrem DJ, Merkow RP, Tomlinson JS, Stewart AK, Ko CY, et al. Application of the pancreatic adenocarcinoma staging system to pancreatic neuroendocrine tumors. J Am Coll Surg. 2007;205(4):558–63 [ Links ]
24. Klimstra DS, Modlin IR, Adsay N, Chetty R, Deshpande V, Goñen M, et al. Pathology reporting of neuroendocrine tumors: application of the Delphic Consensus Process to the development of a minimum pathology data set. Am J Surg Pathol. 2010;34(3):300–13. DOI: 10.1097/ PAS.0b013e3181ce1447. [ Links ]
25. Rindi G, Falconi M, Klersy C, Albarello L, Boninsegna L, Buchler M, et al. TNM staging of neoplasms of the endocrine pancreas: results from a large international cohort study. J Natl Cancer Inst. 2012;104:764–7. DOI: 10.1093/jnci/djs208. [ Links ]
26. Ito T, Igarashi H, Jensen RT. Therapy of metastatic pancreatic neuroendocrine tumors (pNETs): recent insights and advances. J Gastroenterol. 2012;47:941–60. DOI: 10.1007/s00535-012- 0642-8. [ Links ]
27. Kulke MH, Bergsland EK, Yao JC. Glycemic control in patients with insulinoma treated with everolimus. N Engl J Med. 2009;360:195–7. DOI: 10.1056/ NEJMc0806740. [ Links ]
28. Dimou AT, Syrigos KN, Saif MW. Neuroendocrine tumors of the pancreas: what´s new. Highlights from the “2010 ASCO Gastrointestinal Cancers Symposium, Orlando, FL. USA. January 22- 24.2010. JOP (online). 2010;11(2):135–8. [ Links ]
Artículo recibido el 13 de octubre de 2015 y aceptado para publicación el 15 de diciembre de 2015.
Fuente de financiamiento: autofinanciamiento.
Conflictos de intereses: no existen.
Correspondencia: Fernando Revoredo Rego
Av. Grau 800. La Victoria. Lima
Telf. 3242983 anexo 44123
Correo electrónico: fernandorevoredo@hotmail.com
