










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. vol.78 no.4 Lima oct./dic. 2017
http://dx.doi.org/10.15381/anales.v78i4.14262
http://dx.doi.org/10.15381/anales.v78i4.14262
ARTÍCULO ORIGINAL
Detección de la mutación E8SJM en el gen LIPA, por PCR en tiempo real, para la investigación de la enfermedad por almacenamiento de ésteres de colesterol
Detection of the E8SJM mutation in the LIPA gene, by real-time PCR, for the investigation of cholesteryl ester storage disease
Diana Rojas Málaga1, Ursula Matte1,3, Carlos Thadeu Schmidt Cerski2, Roberto Giugliani3,4
1 Centro de Terapia Génica, Hospital de Clínicas de Porto Alegre, RS, Brasil.
2 Servicio de Patología, Hospital de Clínicas de Porto Alegre, RS, Brasil.
3 Departamento de Genética, UFRGS, Porto Alegre, RS, Brasil
4 Servicio de Genética Médica, Hospital de Clínicas de Porto Alegre, RS, Brasil.
Resumen
Introducción: La enfermedad por Almacenamiento de Ésteres de Colesterol (CESD; Cholesteryl Ester Storage Disease) es una enfermedad de depósito lisosomal, su presentación es bastante variable y su diagnóstico constituye un desafío. Además, existe un número de anomalías observadas en los pacientes con CESD que se sobreponen a diagnósticos más comunes, siendo probable que sea subdiagnosticada. La mayoría de pacientes relatados hasta el momento son portadores de la mutación E8SJM en el gen LIPA. En este sentido, el auxilio en el diagnóstico es fundamental pues existen opciones de terapia en desarrollo. Diseño: Estudio observacional. Objetivo: Estandarizar la técnica de PCR en tiempo real para la detección de la mutación más frecuente, E8SJM, en muestras de sangre periférica y de biopsia hepática para el auxilio diagnóstico de CESD y futuros estudios de prevalencia de la mutación. Institución: Centro de Terapia Génica del Hospital de Clínicas de Porto Alegre (HCPA), Brasil. Material biológico: Muestras de ADN extraídas de sangre periférica y tejido hepático parafinado. Principales medidas de resultados: Presencia/Ausencia de la mutación E8SJM. Resultados: Se estandarizó la reacción de PCR en tiempo real, la mutación fue detectada correctamente y posteriormente validada por secuenciación de Sanger. La mutación fue analizada en 137 muestras y encontrada en apenas una paciente que ingresó al Servicio de Genética Médica del HCPA con diagnóstico clínico y bioquímico de CESD/Wolman. Conclusiones: La técnica de PCR en tiempo real es ideal para la detección rápida y en gran escala de la mutación frecuente asociada a CESD.
Palabras clave: Enfermedades por Almacenamiento Lisosomal; Esterol Esterasa; Enfermedad de Acumulación de Colesterol Éster; Enfermedad de Wolman; Técnicas de Diagnóstico Molecular.
Abstract
Introduction: Cholesteryl Ester Storage Disease (CESD) is a lysosomal storage disorder, its presentation is highly variable and its diagnosis challenging. In addition, there are several abnormalities observed in patients with CESD who overlap with more common diagnoses and are likely to be underdiagnosed. Most patients reported to date are carriers of the E8SJM mutation in the LIPA gene. In this sense, diagnostic assistance is essential because there are options for therapy in development, as well as mutation prevalence studies. Design: Observational research. Objective: To standardize the real-time PCR technique for the detection of the most frequent mutation, E8SJM, in peripheral blood and liver biopsy specimens for the diagnosis of CESD and future mutation prevalence studies. Institution: Center of Gene Therapy of the Hospital de Clinicas de Porto Alegre (HCPA), Brazil. Biological material: DNA samples extracted from peripheral blood and paraffinembedded liver tissue. Main outcome measures: Presence / Absence of E8SJM mutation. Results: The PCR reaction was standardized in real time; the mutation was correctly detected and validated by Sanger sequencing. The mutation was analyzed in 137 samples and found in only one patient who entered the Medical Genetics Service of the HCPA with clinical and biochemical diagnosis of CESD/Wolman. Conclusions: The real-time PCR technique is ideal for rapid and large-scale detection of the frequent CESD-associated mutation.
Keywords: Lysosomal Storage Diseases; Sterol Esterase; Cholesterol Ester Storage Disease; Wolman Disease; Molecular Diagnostic Techniques.
INTRODUCCIÓN
La enfermedad por Almacenamiento de Ésteres de Colesterol (CESD; OMIM 278000) es una enfermedad rara, de herencia autosómica recesiva, que pertenece al grupo de enfermedades por depósito lisosomal. La CESD es causada por mutaciones en el gen LIPA (10q23.2 23.3) que afectan la actividad enzimática de la enzima lipasa ácida lisosómica (LAL, EC 3.1.1.13) (1). Este gen, compuesto por 10 exones, codifica una proteína de 399 aminoácidos. Hasta el momento fueron descritas más de 30 mutaciones (de sentido erróneo, sin sentido, en sitios de splicing, pequeñas y grandes deleciones y rearreglos complejos (2) (Figura 1).
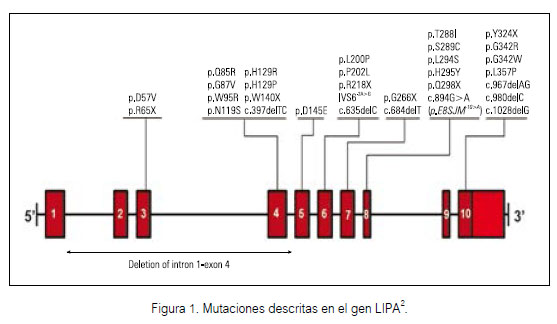
La enzima LAL es responsable por la hidrólisis intracelular de triglicéridos y de ésteres de colesterol derivados de las lipoproteínas plasmáticas (3). Su deficiencia lleva a la acumulación progresiva de estas moléculas en varios órganos y subsecuentemente causa enfermedad hepática, niveles elevados de transaminasas y de esteres de colesterol LDL en el suero. Ocurre hepatomegalia, causado por esteatosis hepática y fibrosis, que pueden llevar a cirrosis micronodular y muerte (4).
La presentación clínica de la CESD es bastante variable. Mientras que algunos pacientes presentan enfermedad hepática en la infancia, otros no son diagnosticados hasta que las complicaciones de manifiestan en la edad adulta (5).
El diagnóstico de la CESD es un desafío. En ocasiones la hepatomegalia es el signo principal, y a veces, el único signo clínico. En el caso de que se realice una biopsia hepática, se observa la combinación de acumulación de macrófagos con evidencia microscópica de almacenamiento lipídico intralisosomal (5). Bioquímicamente, la enfermedad es reconocida por una marcada reducción en la actividad de la lipasa ácida lisosomal (con actividad residual debajo de 10% de lo normal) (6,7). Molecularmente, la enfermedad es confirmada por análisis del gen LIPA y la detección de variantes patogénicas. La principal técnica utilizada es la del PCR convencional donde cada uno de los 10 exones es amplificado y secuenciado utilizando el método de Sanger (4).
Un factor complicador es el hecho de que algunas características clínicas, radio-lógicas y bioquímicas de la CESD son comunes en pacientes que presentan otras enfermedades hepáticas, incluyendo la enfermedad hepática grasa no alcohólica (NAFLD, nonalcoholic fatty liver disease), la esteatosis hepática no alcohólica (NASH, nonalcoholoic steatohepatitis) y la cirrosis criptogénica, entre otras (8-11). Por lo tanto, es probable que los pacientes con CESD sean erróneamente diagnosticados, o no sean diagnosticados. La prevalencia estimada de CESD en la población caucásica e hispánica es de ~0.8 cada 100 000 (~1 en 130 000; 95% CI: ~1 en 90 000 a 1 en 170 000) (12,13).
La grande mayoría de pacientes con CESD, aproximadamente 60% (95% IC: 51%-69%), son portadores de la mutación E8SJM (c.894G>A) en por lo menos un alelo (4,13,14). Esta mutación introduce un sitio de splicing alternativo, resultando en la deleción del exon 8 en el mRNA y originando una proteína 24 aminoácidos más corta. Esta observación justifica la utilización de la detección de esta mutación como un procedimiento inicial en el proceso de genotipaje y hasta mismo como herramienta auxiliar en el proceso diagnóstico. El desarrollo de la terapia de reposición enzimática para la CESD hace que el diagnóstico necesite ser rápido, específico, siendo de extrema importancia, una vez que la terapia parece cambiar substancialmente la historia natural de la enfermedad (15,16).
Sin embargo, antes de que sea aplicable, es necesario la estandarización de las técnicas implicadas, desde el procesamiento de las muestras hasta el análisis e interpretación de los resultados.
MÉTODOS
Material Biológico
Fueron utilizados: 1) noventa y seis muestras de sangre periférica (EDTA) de individuos sanos como controles normales, 2) una muestra positiva para la mutación E8SJM, extraída de sangre periférica y 3) cuarenta muestras de tejido hepático parafinado proveniente de pacientes sometidos a trasplante hepática en el Hospital de Clínicas de Porto Alegre, con diagnóstico prévio de NASH o de cirrosis criptogénica. 4) Además, fue incluida en el análisis una muestra de ADN extraída de sangre periférica de una paciente que entró al Servicio de Genética Médica del HCPA (SGM-HCPA) con diagnóstico clínico y bioquímico de CESD/Wolman. La paciente, de 3 años de edad, presentaba hepatomegalia, esteatosis hepática y en los exámenes laboratoriales un discreto aumento de aminotransferasas y aumento moderado de colesterol y triglicéridos. Los resultados histológicos y de microscopia electrónica sugerían enfermedad de Wolman o de Depósito de ésteres de colesterol (CESD). Bioquímicamente se detectó una actividad enzimática de la lipasa ácida en leucocitos disminuida: 1,1nmol/h/mg prot (valor de referencia:112-378).
MÉTODOS
La extracción de ADN a partir de sangre periférico fue realizada con el kit comercial Easy-DNA® (Invitrogen, USA), de acuerdo a las indicaciones del fabricante. La extracción de DNA a partir de tejido hepático parafinado fue realizada utilizando el método de Coura et al (2005). Los ADN obtenidos fueron cuantificados y la pureza fue analizada en el espectro-fotómetro NanoDrop® ND-1000 (Thermo Scientific, USA).
La identificación de la mutación frecuente, E8SJM, fue realizada por PCR en tiempo real utilizando el sistema de genotipaje Taqman® SNP, utilizando sondas y cebadores personalizados: Cebadores: 5’-CTG GAA CTT CTG TGC AAA ACA TGT3’ y 5’-CCC CAA ATG CAC TCC TGG AA-3’; Sonda Taqman 1 para la secuencia normal: VIC/5’-TGG AGC CAG GTA GGC-3’/ NFQ; Sonda Taqman 2 para la mutación E8SJM: FAM/5’-TGG AGC CAA GTA GGC3’/NFQ) y el StepOnePlusTM Real-Time PCR System (Applied Biosystems, USA). La sonda Taqman de la secuencia normal y de la mutación E8SJM fueron marcadas con los fluorocromos VIC (verde) y FAM (azul), respectivamente.
La reacción de PCR en tiempo real fue realizada siguiendo las instrucciones del fabricante, con algunas modificaciones, utilizando 100ng de ADN por reacción, cada muestra fue analizada en triplicado. El programa de amplificación incluye lo siguiente (ver Tabla 1).

La colecta de datos es realizada a medida que la reacción de PCR ocurre, donde es detectada la señal fluorescente producida proporcionalmente durante la amplificación del ADN blanco. Los productos de PCR fueron analizados utilizando el gráfico de multicomponentes proporcionado por el StepOnePlusTM software (v2.3). El análisis de los datos se traduce en la evaluación de las curvas de amplificación (Figura 2A), en las que se representa la fluorescencia detectada versus el número de ciclos de PCR. Los resultados fueron expresados en ausencia/presencia de la mutación.
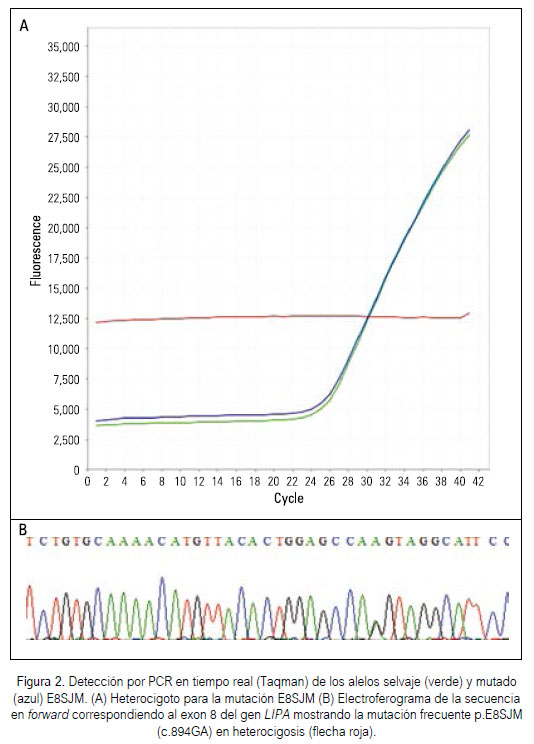
Para la confirmación del alelo positivo para la mutación E8SJM en el PCR en Tiempo Real, la secuenciación se realizó por el método de Sanger del exon 8 utilizando los primers forward 5’-TTAGTGCTTTGAAGGGCAAAA-3’ y reverse 3’-TCTATTTGGAAAGGGTTTGCAT-5’, diseñados utilizando el software online Primer3 v.0.4.0. No se utilizó ningún paquete estadístico pues el presente estudio es un análisis cualitativo de ausencia/presencia de mutación.
Consideraciones Éticas
La presente investigación fue aprobada por sus aspectos éticos y metodológicos por el Comité de Ética en Investigación (CEP) del Hospital de Clínicas de Porto Alegre (proyecto 13-0189).
RESULTADOS
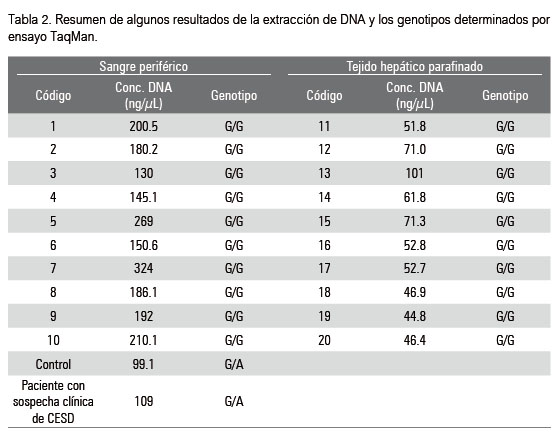
La extracción de ADN a partir de sangre periférica fue realizada con éxito. Se obtuvo un promedio de 205 ng/uL con razón de pureza (DO 260/280) de 1.85. Originalmente el protocolo propuesto por Coura et al (2005) fue utilizado para la extracción de ADN a partir de tejido de carcinoma colorrectal parafinado, esta técnica se caracteriza por ser una alternativa simple y de bajo costo. Esta técnica resultó ser reproducible en nuestra investigación, permitiéndonos extraer ADN con cantidad y calidad suficiente para la realización de los análisis moleculares. Para el ADN extraído de tejido parafinado se obtuvo una concentración promedio de 54ng/uL con razón de pureza (DO 260/280) de 1.88. El detalle de las concentraciones de algunas muestras de DNA analizadas en este estudio se encuentra en la Tabla 2.
La Figura 2A muestra el gráfico de la cinética de amplificación por PCR en tiempo real del DNA extraído a partir de sangre periférica. Durante los ciclos iniciales de la reacción de PCR no se presentó ningún cambio significativo de la intensidad de la fluorescencia emitida. La referencia pasiva (línea roja) permaneció constante a lo largo del proceso de PCR. Se observó que el número de ciclos necesarios para comenzar a obtener señal de fluorescencia detectable fue en promedio 26 (valor Ct). En el control positivo, se observa la amplificación del alelo normal (verde) y del alelo mutado (azul), correspondiendo a un heterocigoto para la mutación E8SJM. Para el ADN extraído del tejido hepático parafinado, el valor Ct fue en promedio 28. No fue detectada la mutación E8SJM en ninguna de las muestras de ADN extraídas de tejido parafinado de pacientes con diagnóstico de NASH o cirrosis criptogénica. La mutación tampoco fue detectada en ninguna muestra de losncontroles normales. Esta mutación fue detectada en la paciente de 3 años que entró al SGM-HCPA con sospecha clínica de CESD (Figura 2A).
Debido a que la secuenciación por el método de Sanger ser considerado el "gold estándar" para el diagnóstico molecular de mutaciones, fue realizado la secuenciación del exon 8 para la confirmación del resultado obtenido por el PCR en tiempo real, confirmándose el estado en heterocigosis de la mutación E8SJM (Figura 2B).
DISCUSIÓN
La enfermedad por Almacenamiento de Ésteres de Colesterol (CESD) es una enfermedad genética rara. Aunque hasta la fecha se han descrito aproximadamente 155 casos en el mundo, es probable que la enfermedad sea más frecuente y las formas menos graves estén siendo subdiagnosticadas (4,17,18,19).
La dificultad de su diagnóstico radica en la heterogeneidad de la enfermedad y en la ausencia de síntomas específicos, los cuales se pueden sobreponer con las de otras enfermedades (20,21). Un estudio reveló la presencia de casos de CESD en grupos de pacientes con hipercolesterolemia, de 276 individuos analizados, 5 presentaron una variante patogénica en el gen LIPA, existiendo 2 heterocigotos para E8SJM (22). Un segundo trabajo, con un objetivo similar, tamizó la mutación E8SJM en pacientes con elevación aislada de LDL, hiperlipidemia combinada, deficiencia de HDL y dislipidemia mixta; de los 1375 pacientes analizados, 6 individuos presentaron la mutación E8SJM, siendo 3 de estos, heterocigotos compuestos y así afectados con CESD (23). La identificación de portadores y afectados en estas poblaciones, implica que la misma está enriquecida con portadores de mutaciones en el gen LIPA y subraya la importancia de las pruebas genéticas para las enfermedades raras en las cuales las manifestaciones clínicas pueden superponerse al fenotipo de trastornos más prevalentes. A pesar de la gran semejanza en las manifestaciones clínicas entre estas enfermedades hepáticas NAFLD, NASH, cirrosis criptogénica y CESD, no se ha publicado en la literatura algún trabajo donde haya sido estudiado una cohorte de pacientes, inicialmente diagnosticados con estas enfermedades con el fin de excluir CESD, confirmando así la hipótesis diagnóstica inicial.
Diferentes estudios han utilizado distintas fuentes para obtener el ADN para la detección de mutaciones en el gen LIPA (leucocitos extraídos de sangre periférica, sangre impregnada en papel filtro, fibroblastos, saliva, entre otros), así como diferentes metodologías para la detección de las variantes en el gen (MS-PCR, secuenciación por Sanger y RT-PCR) (4,12). Existen otras técnicas moleculares modernas, entre ellos la secuenciación de nueva generación utilizando paneles de genes o secuenciación del exoma, que presentan varias ventajas como la rapidez de resultados y la posibilidad de analizar varios en paralelo; sin embargo, el costo de estos análisis hace inviable su utilización como herramienta de tamizaje (24). En este trabajo proponemos el PCR en tiempo real como una herramienta para el tamizaje a gran escala de la mutación E8SJM (presente en 60% de los pacientes), asociada a la CESD (13). En este escenario, nuestra investigación trae como novedad la utilización de ADN extraído de tejido hepático parafinado usando un protocolo casero de bajo costo, seguido de PCR en tiempo real para la detección de la mutación.
La técnica de PCR en tiempo real, con la utilización de las sondas Taqman, fue estandarizada y permitió determinar rápidamente la presencia o ausencia de la alteración génica más común. A pesar de que la mutación E8SJM no fue detectada en ninguna de las muestras analizadas, la utilidad clínica de este ensayo fue demostrada con el análisis molecular de la paciente con diagnóstico clínico y bioquímico de CESD/Wolman. La detección de la mutación por PCR en tiempo real fue realizada de manera exitosa, encontrándose en estado de heterocigosis, lo cual significa que la paciente posee una copia del gen mutado y confirmado por secuenciación de Sanger (Figura 2B).
El PCR en tiempo real representa una técnica simple, rápida y confiable para un tamizaje inicial en el diagnóstico de CESD, pudiendo incluso ser utilizada en estudios de prevalencia de CESD. Contar con esta herramienta para el auxilio de tamizaje a gran escala de esta mutación es fundamental desde que no existen datos de prevalencia en Latinoamérica.
Referencias Bibliográficas
1. Anderson RA, Bryson G M, Parks J S. Lysosomal acid lipase mutations that determine phenotype in Wolman and cholesterol ester storage disease. Molecular Genetics and Metabolism.1999;68(3):333–345. https://doi.org/10.1006/mgme.1999.2904. [ Links ]
2. Marin-Valencia I, Pascual J. Cap 36 Wolman Disease. Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric. Elsevier. Fifth Edition. 2015: 403- 410.
3. Human Genome Mutation Database (HGMD), consultado 23/11/2016. Disponible en: http://www.hgmd.cf.ac.uk/ac/index.php.
4. Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease. Journal of Hepatology. 2013; 58(6):1230–1243. https://doi.org/10.1016/j.jhep.2013.02.014. [ Links ]
5. Aguisanda F, Thorne N, Zheng W. Targeting Wolman Disease and Cholesteryl Ester Storage Disease: Disease Pathogenesis and Therapeutic Development. Current Chemical Genomics and Translational Medicine. 2017; 11: 1–18. https://doi.org/ 10.2174/2213988501711010001. [ Links ]
6. Weiler C, Freudenberg F, Müller-Höcker J. Cholesterol ester storage disease-A rare disease or a rare diagnosis?. Pathologe. 2009; 30(1): 65-69. https://doi.org/ 10.1007/s00292-009-1124-5. [ Links ]
7. Boldrini, Devito R, Biselli R, Filocamo M, Bosman C. Wolman disease and cholesteryl ester storage disease diagnosed by histological and ultrastructural examination of intestinal and liver biopsy. Pathology Research and Practice. 2004;200(3):231-240. https://doi.org/10.1016/j.prp.2003.11.001. [ Links ]
8. Hulkova H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology.2012;60 (7): 1107-13. https://doi. org/10.1111/j.1365-2559.2011.04164.x. [ Links ]
9. Gasche C, Aslanidis C, Kain R, Exner M, Helbich T, Dejaco C, Schmitz G, Ferenci P. A novel variant of lysosomal acid lipase in cholesteryl ester storage disease associated with mild phenotype and improvement on lovastatin. J Hepatol. 1997; 27(4):744-50. https://doi.org/10.1016/S0168-8278(97)80092-X. [ Links ]
10. Chatrath H, Keilin S, Attar B. Cholesterol ester storage disease (CESD) diagnosed in an asymptomatic adult. Digestive diseases and sciences. 2009; 54 (1): 168-73. https://doi.org/10.1007/s10620-008-0310-2. [ Links ]
11. Hamilton J, Jones I, Srivastava R, Galloway P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin. Chim. Acta. 2012; 413:1207–1210. https://doi.org/ 10.1016/j.cca.2012.03.019. [ Links ]
12. Muntoni S, Wiebusch H, Jansen-Rust M, Rust S, Seedorf U, Schulte H, et al. Prevalence of Cholesteryl Ester Storage Disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(8): 1866–1868. https://doi.org/10.1161/ATVBAHA.107.146639. [ Links ]
13. Scott S, Liu B, Nazarenko I, Martis S, Kozlitina J, Yang Y, et al. Frequency of the Cholesteryl Ester Storage Disease Common LIPA E8SJM Mutation (c.894G>A) in Various Racial and Ethnic Groups. Hepatology. 2013; 58(3): 958-965. https://doi.org/10.1002/hep.26327. [ Links ]
14. Fasano T, Pisciotta L, Bocchi L, Guardamagna O, Assandro P, Rabacchi C, et al. Calandra S. Lysosomal lipase deficiency: Molecular characterization of eleven patients with Wolman or cholesteryl ester storage disease. Molecular Genetics and Metabolism. 2012; 105(3): 450–456. https://doi.org/10.1016/j.ymgme.2011.12.008.
15. Balwani M, Breen C, Enns GM, Deegan PB, Honzík T, Jones S, et al. Clinical effect and safety profile of recombinant human lysosomal acid lipase in patients with cholesteryl ester storage disease. Hepatology. 2013; 58(3): 950–957. https://doi.org/10.1002/hep.26289. [ Links ]
16. Burton BK, Balwani M, Feillet F, Barić I, Burrow TA, Camarena C, et al. A Phase 3 Trial of Sebelipase Alfa in Lysosomal Acid Lipase Deficiency. New England Journal of Medicine. 2015; 373(11): 1010–1020. https://doi.org/10.1056/NEJMoa1501365.
17. Kamenet E, Mollaki V, Hamilton J, Zakharova E & Drogari E. Molecular analysis of 15 homozygous LAL-D patients from Greece using DBS screening. Atherosclerosis. 2017; 263: e100-e101. https://doi.org/10.1016/j.atherosclerosis.2017.06.328 [ Links ]
18. Carbajal-Rodríguez L. Deficiencia de lipasa ácida lisosomal. Reporte de dos casos. Revista Mexicana de Pediatria. 2016; 83 (2): 49-54 [ Links ]
19. Reynders J, Burton BK, del Angel G. Novel LIPA mutations resulting in lysosomal acid lipase deficiency. Molecular Genetics and Metabolism. 2017; 20 (1-2): S114–S115. https://doi.org/10.1016/j. ymgme.2016.11.294 [ Links ]
20. Bernstein D, Desnick R. Cholesteryl ester storage disease (CESD): An under-recognized and treatable LSD with liver dysfunction/failure and dyslipidemia. Molecular Genetics and Metabolism. 2013; 108(2): S24. https://doi.org/10.1016/j.ymgme.2012.11.036 [ Links ]
21. Basu P, Nair T, Farhat S, Foustin S, Ang L, Anwarullah A, et al. Unrevealing a novel association of cholesterol ester storage disease (CESD) & nonalcoholic fatty liver disease(NAFLD)-a similar clinical spectrum with different etiology a prospective clinical study. Journal of Hepatology. 2012; 56(2): S508. https://doi.org/10.1016/S0168-8278(12)61299-9 [ Links ]
22. Sjouke B, Defesche JC, de Randamie JSE, Wiegman A, Fouchier SW, Hovingh GK. Sequencing for LIPA mutations in patients with a clinical diagnosis of familial hypercholesterolemia. Atherosclerosis. 2016; 251: 263-265. http://dx.doi.org/10.1016/j.atherosclerosis.2016.07.008 [ Links ]
23. Kane JP, Stock E, Pullinger CR, Movsesyan I, Malloy MJ, Quinn A. Identifying Cases of Cholesteryl Ester Storage Disease in a Tertiary Lipid Clinic. Journal of Clinical Lipidology. 2013; 7(3): 249. http://dx.doi.org/10.1016/j.jacl.2013.03.034 [ Links ]
24. Stitziel NO, Fouchier SW, Sjouke B, Peloso GM, Moscoso AM, Auer PL, et al. Blood Institute GO Exome Sequencing Project. Exome sequencing and directed clinical phenotyping diagnose cholesterol ester storage disease presenting as autosomal recessive hypercholesterolemia. Arteriosclerosis, thrombosis, and vascular biology. 2013; 33(12): 2909-14. https://doi.org/10.1161/ATVBAHA.113.302426 [ Links ]
Correspondencia
Diana Rojas Málaga dmalaga@hcpa.edu.br
Telef. +55 51 3359-7212 Direcc. Servicio de Genética Médica, Hospital de Clínicas de Porto Alegre. Rua Ramiro Barcelos 2350, Porto Alegre RS 90035-903 Brasil.
Conflicto de intereses: No existe conflicto de interés en el presente trabajo.
Financiamiento: Este trabajo fue financiado por FIPE/HCPA (proyecto 13-0189)
Consentimiento:
Todos los experimentos presentados en esta publicación están incluidos en el proyecto de investigación aprobado por sus aspectos éticos y metodológicos por el Comité de Ètica en investigación del Hospital de Clínicas de Porto Alegre (proyecto 13-0189).
Recibido: 25 de mayo 2017
Aceptado: 2 de septiembre 2017.

