










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkAnales de la Facultad de Medicina
Print version ISSN 1025-5583
An. Fac. med. vol.79 no.1 Lima Jan./Mar. 2018
http://dx.doi.org/10.15381/anales.v79i1.14598
GUÍA DE PRÁCTICA CLÍNICA
Guía de práctica clínica para el diagnóstico y tratamiento de hemofilia en el Seguro Social de Salud del Perú (EsSalud)
Clinical practice guideline for the diagnosis and treatment of hemophilia for the Peruvian Social Security (EsSalud)
Saúl Mendoza Ordoñez 1, Nancy Loayza Urcia 1, Maribel Trujillo Cerna 2, Celina Herrera Cunti 2,Rommel Yanac Avila 3, Walter Ormeño Apaza 3, Carlos Delgado Silva 4, David Díaz Robles 4, Adrián V. Hernández 5,6, Alejandro Piscoya 6,7, Víctor Suárez Moreno 8, Raúl Timaná-Ruiz 8
1 Sociedad Peruana de Hematología. Lima, Perú.
2 Hospital Nacional Guillermo Almenara Irigoyen, EsSalud. Lima, Perú.
3 Hospital Nacional Alberto Sabogal Sologuren, EsSalud. Lima, Perú.
4 Hospital Nacional Dos de Mayo, Ministerio de Salud. Lima, Perú.
5 University of Connecticut/Hartford Hospital Evidence-based Practice Center. Hartford, USA.
6 Escuela de Medicina, Universidad Peruana de Ciencias Aplicadas. Lima, Perú.
7 Hospital Guillermo Kaelin de la Fuente, EsSalud. Lima, Perú.
8 Instituto de Evaluación de Tecnologías en Salud e Investigación, EsSalud. Lima, Perú.
RESUMEN
Introducción. La hemofilia es un trastorno hemorrágico congénito poco común que requiere un manejo interdisciplinario, complejo, y frecuentemente costoso. El objetivo de la presente guía de práctica clínica (GPC) es proveer recomendaciones clínicas basadas en evidencia para el diagnóstico y tratamiento de hemofilia en el seguro social del Perú (EsSalud). Métodos. Se conformó un grupo elaborador local (GEG-Local) conformado por especialistas en hematología y metodólogos. El GEG-Local formuló ocho preguntas clínicas a ser respondidas por la presente GPC. Se buscaron y seleccionaron GPC de hemofilia que respondieran a las preguntas planteadas y obtuvieran un puntaje mayor a 60% en los dominios uno y tres del instrumento Appraisal of Guidelines for Research and Evaluation II (AGREE-II). Durante el 2016 se realizaron búsquedas bibliográficas en Pubmed, EMBASE y biblioteca Cochrane, para actualizar siete preguntas clínicas de la GPC preseleccionada, y para responder una pregunta de novo. En reuniones de trabajo periódicas, el GEG-Local revisó la evidencia y formuló las recomendaciones y flujogramas usando la metodología Grading of commendations Assessment, Development, and Evaluation (GRADE). Finalmente, la GPC fue aprobada con Resolución N° 32-IETSI-ESSALUD-2016. Resultados. La presente GPC abordó ocho preguntas clínicas. En base a dichas preguntas se formularon 22 recomendaciones (tres recomendaciones fuertes y 19 recomendaciones condicionales) y cuatro flujogramas. Conclusión. Este artículo es el resumen de la GPC de EsSalud, en la cual se valoró la evidencia científica disponible sobre diagnóstico y tratamiento de hemofilia.
Palabras clave: Hemofilia; Guía de Práctica Clínica; Medicina Basada en Evidencias (fuente: DeCS Bireme).
ABSTRACT
Introduction. Hemophilia is a rare congenital bleeding disorder; which requires interdisciplinary, complex, and often expensive management. The objective of this clinical practice guideline (CPG) is to provide evidence-based clinical recommendations for the evaluation and management of patients with hemophilia in the Peruvian Social Security (EsSalud). Methods. A local elaboration group (GEG-Local) was established, conformed by specialists in hematology and methodologists. The GEG-Local formulated 8 clinical questions to be answered by this CPG. We searched for and selected hemophilia CPGs that answered the questions posed and obtained a score higher than 60% in domains 1 and 3 of the Appraisal of Guidelines for Research and Evaluation II (AGREE-II). During 2016, bibliographic searches were conducted in Pubmed, EMBASE and the Cochrane library, to update 7 clinical questions of the preselected CPG, and to answer a question de novo. In regular work meetings, the GEG-Local reviewed the evidence and formulated the recommendations and flowcharts using the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) methodology. Finally, the CPG was approved with Resolution No. 32-IETSI-ESSALUD-2016. Results. This CPG addressed 8 clinical questions. Based on these questions, 22 recommendations were formulated (three strong recommendations and 19 weak recommendations) and four flowcharts. Conclusion. This article is the summary of the EsSalud CPG, in which the available scientific evidence on the diagnosis and treatment of hemophilia was assessed.
Keywords: Hemophilia; Practice Guideline, Evidence-Based Medicine (source: MeSH NLM).
INTRODUCCIÓN
La hemofilia es un trastorno hemorrágico congénito, poco común, complejo de diagnosticar y tratar. Esta enfermedad está vinculada a mutaciones en el cromosoma X, que causan deficiencia del factor VIII de coagulación (FVIII) (hemofilia A) o del factor IX (FIX) (hemofilia B) (1). La consecuencia principal de esta enfermedad es la discapacidad, y con ello el deterioro progresivo de la calidad de vida a una edad muy temprana.
La hemofilia requiere un manejo interdisciplinario, complejo, y frecuentemente costoso. Por ello, la Dirección de Guías de Práctica Clínica, Farmacovigilancia y Tecnovigilancia del Instituto de Evaluación de Tecnologías en Salud e Investigación (IETSI) del Seguro Social de Salud del Perú (EsSalud) elaboró una guía de práctica clínica (GPC) basada en evidencias para el diagnóstico y tratamiento de hemofilia (aprobada con Resolución N° 32-IETSIESSALUD-2016), cuyas recomendaciones se encuentran detalladas en la tabla 1 y serán aplicadas por profesionales de la salud en todos los niveles de atención de EsSalud. El presente artículo es un resumen de dicha GPC.
METODOLOGÍA
El procedimiento seguido para la elaboración de la presente GPC se encuentra detallado en su versión "in extenso" en: http://www.essalud.gob.pe/ietsi/guias_pract_clini.html.
Conformación del GEG-Local: El grupo elaborador de la guía local (GEG-Local) incluyó metodólogos y médicos especialistas en hematología de EsSalud, del Ministerio de Salud, y de la Sociedad Peruana de Hematología.
Planteamiento de preguntas clínicas y desenlaces: En concordancia con el alcance de esta GPC, el GEG-Local formuló ocho preguntas clínicas, y definió los desenlaces críticos e importantes para cada pregunta clínica. Las preguntas abordadas fueron:
-
Criterios clínicos y de laboratorio para el diagnóstico de hemofilia
-
- Pregunta uno: "¿Cuáles son los criterios clínicos y de laboratorio para sospechar hemofilia?
-
- Pregunta dos: ¿Cuáles son los criterios de confirmación diagnóstica para hemofilia?
-
- Pregunta tres: ¿Cómo se diagnostica sangrado articular agudo en un paciente con artropatía hemofílica crónica?
-
Manejo de los pacientes hemofílicos (adultos y niños) con hemorragias graves que conllevan riesgo vital inmediato
-
- Pregunta cuatro: ¿Cuál debe ser el manejo de los pacientes hemofílicos (adultos y niños) con hemorragias graves que conllevan riesgo vital inmediato?
-
Manejo de los pacientes hemofílicos (adultos y niños) con hemorragias que no conlleven riesgo vital inmediato
-
- Pregunta cinco: ¿Cuál debe ser el manejo de los pacientes hemofílicos (adultos y niños) con hemorragias graves que no conllevan riesgo vital inmediato?
-
Profilaxis en pacientes hemofílicos (A y B) para prevenir daño articular
-
- Pregunta seis: ¿Cuáles son los tratamientos profilácticos de un paciente hemofílico (A y B) para prevenir daño articular?
-
- Pregunta siete: ¿Cuándo se debe iniciar tratamiento profiláctico en pacientes hemofílicos?
-
Manejo de la hemofilia en niños y adultos en cirugía mayor y menor
-
- Pregunta ocho: ¿Cómo debe ser el manejo de la hemofilia, en niños y adultos en cirugía mayor y menor?
Búsqueda y evaluación de GPC: Se buscó GPC de hemofilia que respondieran a las preguntas planteadas. Se seleccionaron las GPC que tuvieron puntajes mayores al 60% en los dominios uno y tres del instrumento Appraisal of Guidelines for Research and Evaluation II (AGREE-II). Solo una GPC alcanzó estos criterios: la Guía Clínica AUGE de Hemofilia del Ministerio de Salud de Chile 2013 (en adelante, GPC de Chile) (2). La GPC de Chile basaba muchas de sus recomendaciones en los consensos de la guía de la Federación Mundial de Hemofilia (1), por lo cual en el desarrollo de las recomendaciones también se hará referencia a esta guía.
Identificación y graduación de la calidad de la evidencia: De las ocho preguntas clínicas planteadas, siete fueron contestadas por la GPC seleccionada, por lo cual se actualizó la evidencia para estas preguntas realizando una búsqueda sistemática de documentos a partir del 2010, que fue cuando se realizó la búsqueda en la GPC seleccionada. Para la pregunta que no fue contestada por dicha GPC (pregunta tres), se realizó una búsqueda bibliográfica de novo. Las búsquedas fueron realizadas en los buscadores Pubmed, EMBASE y biblioteca Cochrane durante el 2016. Los criterios de inclusión de estudios y los términos de búsqueda para cada preguntan se muestran en el material suplementario N° 1. Debido a que para las preguntas planteadas los estudios fueron muy heterogéneos, la calidad de la evidencia no fue evaluada.
Formulación de las recomendaciones: Luego de revisar la evidencia recolectada para cada pregunta, se formuló recomendaciones usando la metodología Grading of Recommendations Assessment, Development, and Evaluation (GRADE) (3). Se tuvo en consideración: 1) Beneficios y daños de las opciones, 2) Valores y preferencias de los pacientes, 3) Aceptabilidad por parte de los profesionales de salud, 4) Viabilidad de las opciones en los establecimientos de salud de EsSalud, y 5) Uso de recursos.
Las recomendaciones pudieron ser consideradas recomendaciones fuertes o recomendaciones condicionales:
-
- Recomendación fuerte (usando el término "se recomienda") cuando el GEG-Local consideró que todos o casi todos los profesionales que revisan la evidencia disponible seguirían esta recomendación.
-
- Recomendación condicional (usando el término "se sugiere") cuando el GEG-Local consideró que la mayoría de los profesionales que revisan la evidencia disponible seguirían esta recomendación, pero un grupo de profesionales podrían no seguirla.
Revisión por expertos externos y pacientes: La presente GPC fue revisada en reuniones con médicos especialistas representantes de otras instituciones (EsSalud, Ministerio de Salud, Sociedad Peruana de Hematología), tomadores de decisiones, y pacientes. Además, fue enviada para su revisión por un experto externo mencionado en la sección de agradecimientos.
Aprobación de la GPC: La presente GPC fue aprobada para su uso en EsSalud, con Resolución N° 32-IETSI-ESSALUD-2016.
Actualización de la GPC: La presente GPC tiene una validez de tres años. Al acercarse el fin de este período, se procederá a realizar una revisión de la literatura para su actualización, luego de la cual se decidirá si se actualiza la presente GPC o se procede a realizar una nueva versión.
RECOMENDACIONES
La presente GPC abordó ocho preguntas clínicas. En base a dichas preguntas se formularon 22 recomendaciones (tres recomendaciones fuertes y 19 recomendaciones condicionales) (Tabla 1) y cuatro flujogramas (Figuras 1, 2, 3 y 4). A continuación, se detallarán las recomendaciones para cada pregunta. El número de cada recomendación abordada está acorde a la numeración de la Tabla 1.
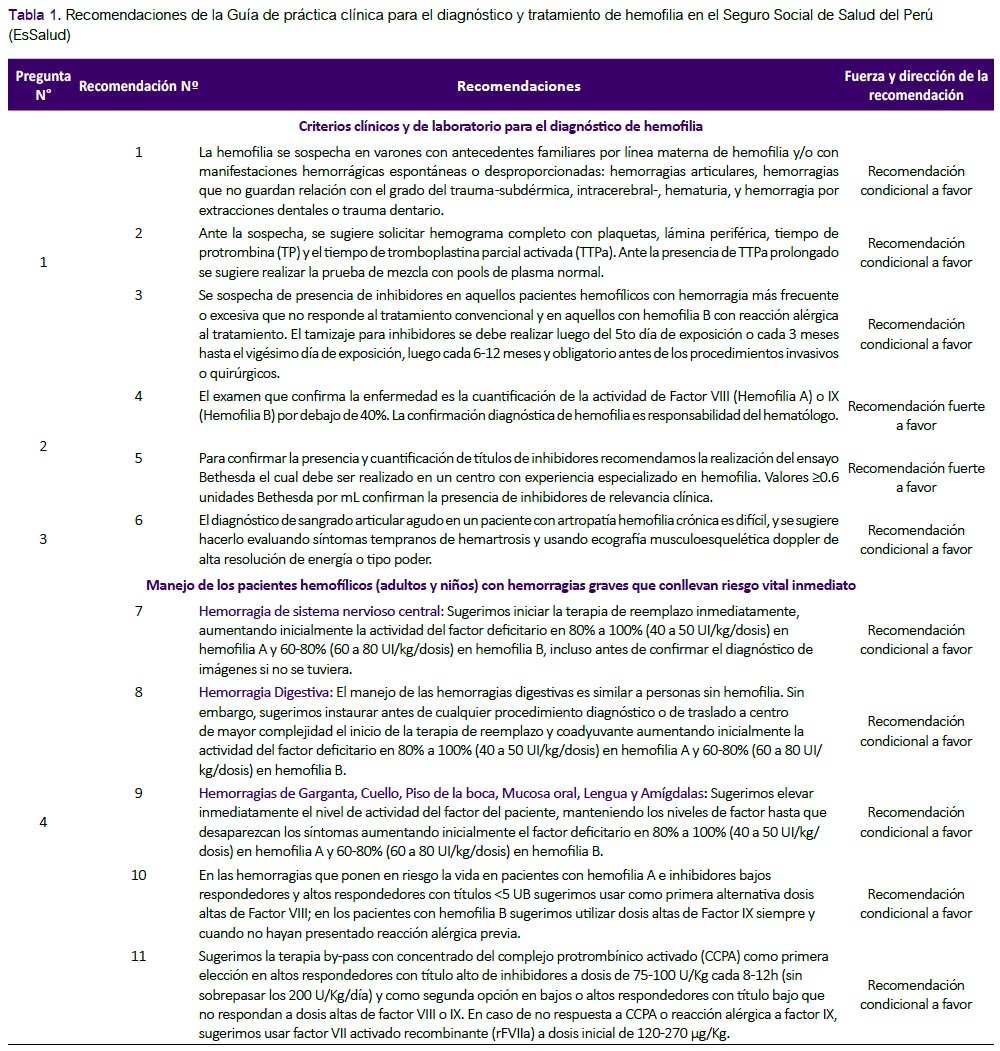
Pregunta 1: ¿Cuáles son los criterios clínicos y de laboratorio para sospechar hemofilia?
Recomendación 1: Se adoptaron los criterios de sospecha clínica que propone la GPC de Chile. Se debe considerar que un 30% de los enfermos no presentan antecedentes familiares conocidos. (4) En recién nacidos a término, los síntomas de sangrado característicos incluyen sangrado intracraneal luego del nacimiento, hinchazón dolorosa de articulaciones causada por hemartrosis, equimosis sin explicación cuando un bebé empieza a gatear o caminar, sangrado postoperatorio, sangrado subcutáneo extenso luego de uso de agujas dérmicas, y sangrado muscular (espontáneo o luego de vacunación intramuscular)(5). Resulta importante que los exámenes de laboratorio sólo se realicen en quienes tengan sospecha clínica, puesto que dichos exámenes implican mayor costo y mayor riesgo debido a la necesidad de punción venosa.
También resulta de importancia realizar la diferenciación entre un defecto hemostático primario (DHP) (ejm: defectos plaquetarios, Enf. von Willebrand o defectos de la pared de los vasos sanguíneos) y una deficiencia de factor de coagulación (DFC) (ejm: hemofilia y desordenes de coagulación), para lo cual sugerimos considerar las siguientes características clínicas (basada en la GPC de Chile) (2):
-
Lugar de sangrado: en el caso de DHP: piel y membranas mucosas; en el caso de DFC: tejidos blandos, músculos y articulaciones
-
Sangrado luego de cortes menores: se dan en el caso de DHP, pero no son usuales en el caso de DFC
-
Petequias: presentes en el caso de DHP, ausentes en el caso de DFC
-
Equimosis: pequeñas y superficiales en el caso de DHP; grandes, profundas y palpables en el caso de DFC
-
Hemartrosis: raras en el caso de DHP, comunes en el caso de DFC
-
Hematomas de tejidos blandos: raras en el caso de DHP, característicos en el caso de DFC
-
Sangrado luego de trauma o cirugía: inmediato en el caso de DHP, retardado en el caso de DFC
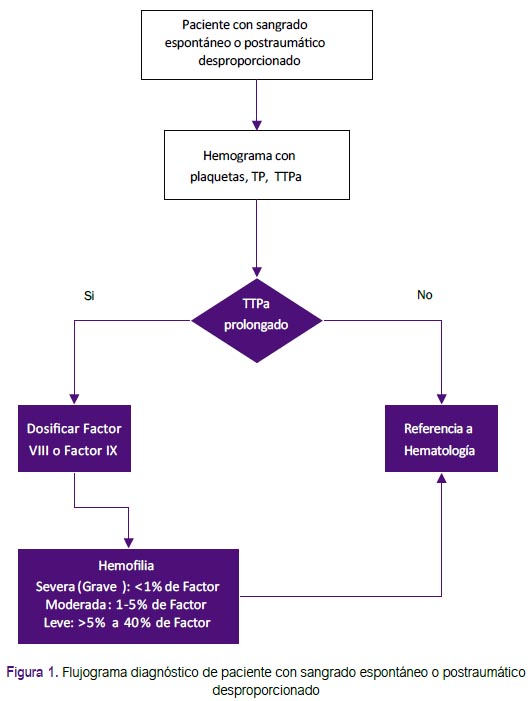
Recomendación 2: Se adoptaron los exámenes de laboratorio que la GPC de Chile (2) y un consenso de especialistas (5) recomendaron considerar ante la sospecha de hemofilia: TTPa, TP, y hemograma con recuento de plaquetas. De manera que, si el TTPa es prolongado y el número de plaquetas es normal, se deberá realizar la prueba de mezcla con pools de plasma normal. Si esta prueba corrige el TTPa, se planteará la sospecha de hemofilia(4). (Figura 1) Además, se resaltó que el tiempo de sangría no sería recomendado debido a que podría generar mayor sangrado, y los estudios de imágenes de rutina no serían necesarios.(1)
Recomendación 3: Los pacientes con hemofilia que reciben reemplazo de factores de coagulación, pueden desarrollar una reacción inmunológica, produciendo anticuerpos contra dichos factores de coagulación exógenos (inhibidores); lo cual impide una adecuada coagulación. (1) Se adoptó la sugerencia de la GPC de Chile de sospechar de presencia de inhibidores en pacientes hemofílicos con hemorragia más frecuente o excesiva que no responde al tratamiento convencional y en aquellos con hemofilia B con reacción alérgica al tratamiento. Asimismo, se adoptó la recomendación de la guía de la Federación Mundial de Hemofilia (1) de que los pacientes sean tamizados para inhibidores luego de haber sido expuestos al anticuerpo durante cinco días; este tamizaje se realizará cada 3 meses hasta el vigésimo día de exposición; y posterior a ello, cada 6-12 meses. Además, se deberá realizar antes de los procedimientos invasivos o quirúrgicos.
Pregunta 2. ¿Cuáles son los criterios de confirmación diagnóstica para hemofilia?
Recomendación 4: Debido a la correlación existente entre la actividad de los factores VIII y IX y las manifestaciones clínicas, la Sociedad Internacional de Trombosis y Hemostasia (ISTH) y la Federación Mundial de Hemofilia recomiendan mantener la clasificación de hemofilia de acuerdo a la actividad de los factores VIII y IX de la siguiente manera: severa (<1%), moderada (entre 1 y 5%), y leve (entre 5 y 40%)(1, 6).
Recomendación 5: Con respecto a los títulos de anticuerpos inhibidores de los factores VIII o IX, la ISTH y la Federación Mundial de Hemofilia recomiendan usar como punto de corte el valor de 0.6 unidades Bethesda para confirmar la presencia de inhibidores(1, 6). Este punto de corte es utilizado debido a que un estudio halló que este punto de corte era preciso para identificar inhibidores cuando se usa el ensayo de Bethesda modificado por Nijmegen(7).
Pregunta 3: ¿Cómo se diagnostica sangrado articular agudo en un paciente con artropatía hemofílica crónica?
Recomendación 6: La hemartrosis (HA) consiste en un derrame agudo de sangre al interior de una o varias articulaciones. Es el evento más frecuente, y representa el 65% a 80% de todas las hemorragias en el paciente hemofílico. Los frecuentes episodios hemorrágicos desencadenan artropatía hemofílica crónica (AHC), que consiste en un proceso degenerativo articular producto del daño secundario de las hemorragias. Esta complicación de la enfermedad puede exacerbarse con un nuevo episodio hemorrágico agudo. En este contexto es difícil diagnosticar las exacerbaciones debido a la artropatía subyacente. Sin embargo, estos sangrados esporádicos son los que producen la disfunción articular a largo plazo(1).
-
Diferenciación por examen clínico: No hay protocolo o criterios validados disponibles para diferenciar HA de AHC. Timmer et al. 2015 (8) hicieron una revisión narrativa para identificar información que diferencie ambos cuadros, encontrando que en nueve estudios de HA, los síntomas tempranos (menor a una hora de presentación) fueron dolor y sensaciones de tensión en la articulación, rigidez y hormigueo; en tanto que los síntomas tardíos incluyeron tumefacción, aumento de temperatura local, rango articular reducido, enrojecimiento, inhabilidad de mover la articulación, sensibilidad, piel brillante, espasmo muscular, y posición fija de la articulación (Material suplementario N° 2)(8).
-
Diferenciación por imágenes: El diagnóstico por imágenes se suele hacer con la ecografía doppler de alta resolución de energía o tipo poder (PDUS), ecografía que usa una mayor amplitud de la señal doppler para detectar la materia en movimiento, como fluidos o sangre; o con resonancia magnética nuclear (RMN). Melchiorre et al. 2013 (9) evaluaron la capacidad de PDUS para detectar sangrado y daño articular en 82 pacientes (103 articulaciones) con hemofilia A o B y AHC en comparación con la evaluación clínica. Los autores encontraron una alta correlación entre el "score US" y el número de sangrados verificados por aspiración (Rho de Spearman = 0,375, p<0,01); concluyendo que PDUS puede ser útil y confiable para evaluar modificaciones de la articulación, incluyendo sangrado. Ceponis et al. 2013 (10) realizaron un estudio observacional y encontraron que solo un tercio de sangrados fueron diagnosticados con examen clínico en comparación a PDUS + aspiración; lo cual resalta la importancia de este examen auxiliar.
Por otro lado, la RMN evalúa adecuadamente las partes blandas en las articulaciones, y permite visualizar cambios tempranos y tardíos de AHC (8). Además, permite ver depósitos de hemosiderina, una huella de un sangrado previo. Sin embargo, no se encontró literatura de RMN que evalúe la diferenciación entre HA y la exacerbación de AHC, además que la disponibilidad y costos elevados del estudio, dificultan su uso rutinario.
Pregunta 4. ¿Cuál debe ser el manejo de los pacientes hemofílicos (adultos y niños) con hemorragia graves que conllevan riesgo vital inmediato?
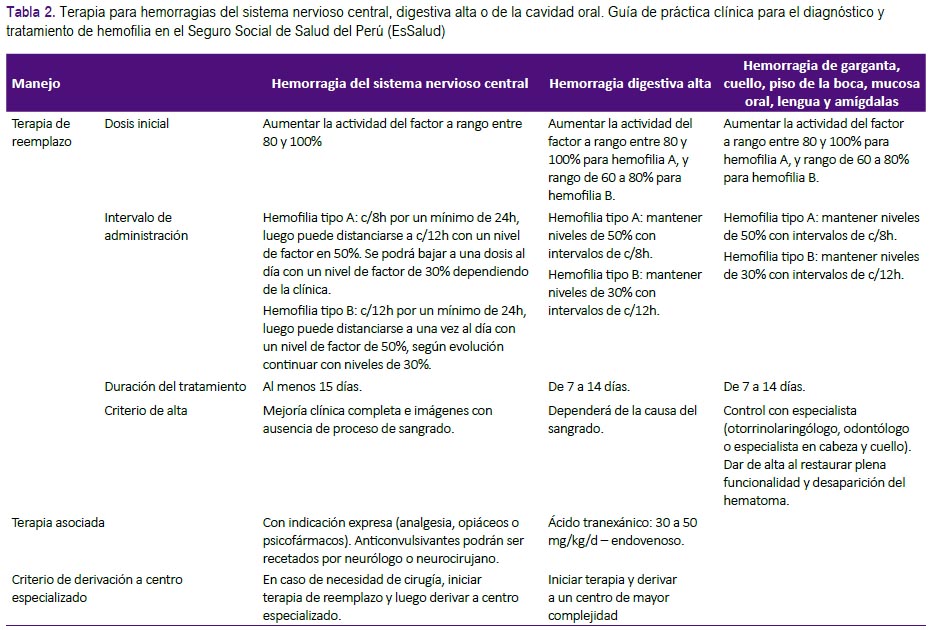
Recomendaciones 7, 8 y 9: El manejo de los pacientes hemofílicos con hemorragia graves que conllevan riesgo vital inmediato resulta de gran importancia, y debería ser estandarizado. Se formula-ron recomendaciones sobre las dosis y la duración del tratamiento con factores de coagulación para hemorragias del sistema nervioso central, hemorragia digestiva alta y hemorragia de la cavidad oral. (Tabla 2 y Figura 2) Estas fueron adoptadas de las recomendaciones de la GPC de Chile, que fueron consistentes con lo recomendado por la Federación Mundial de Hemofilia(1, 2).
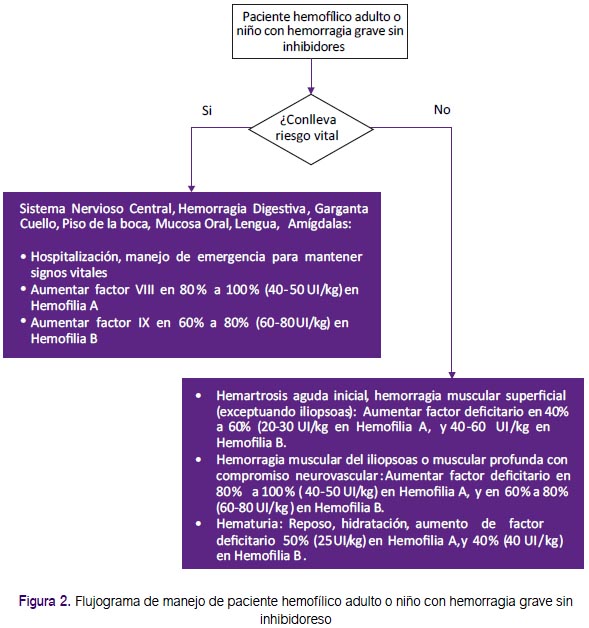
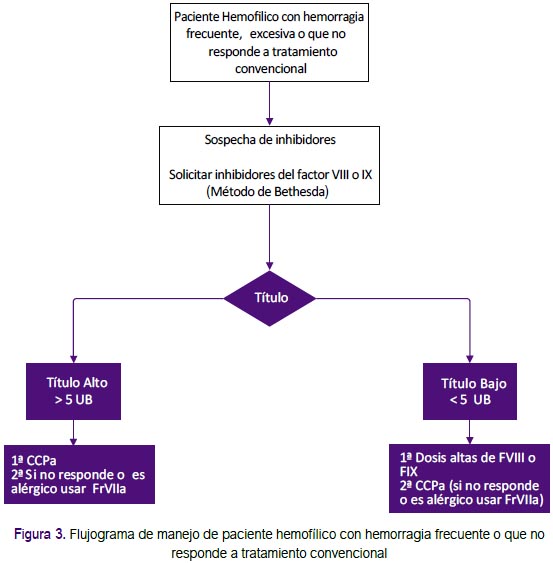
Recomendaciones 10 y 11: Se formularon recomendaciones para el caso de pacientes que presenten inhibidores: Para aquellos bajos respondedores o altos respondedores con títulos <5 unidades Bethesda, se prefiere el uso de factores de coagulación; y para aquellos que no responden a esta terapia se recomienda el uso de terapia bypass. Para pacientes con títulos más altos de inhibidores se recomienda iniciar con terapia bypass (complejo protrombínico activado [CCPA] o factor VII recombinado activado (rFVIIa). Estas fueron adoptadas de las recomendaciones de la GPC de Chile, que fueron consistentes con lo recomendado por el consenso emitido por el Comité Latinoamericano sobre la Terapéutica de Personas con Inhibidores (CLOTTING)(2, 11). (Figura 3)
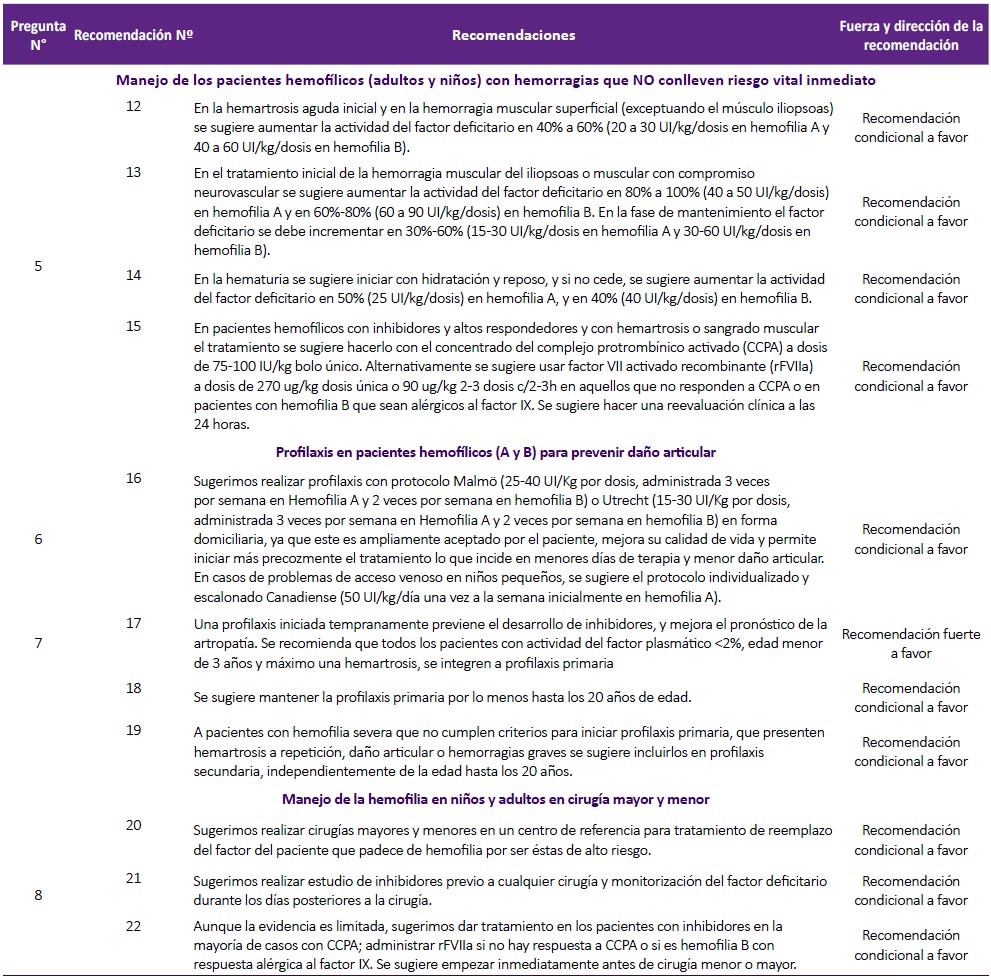
Pregunta 5: ¿Cuál debe ser el manejo de los pacientes hemofílicos (adultos y niños) con hemorragias que NO conlleven a riesgo vital inmediato?
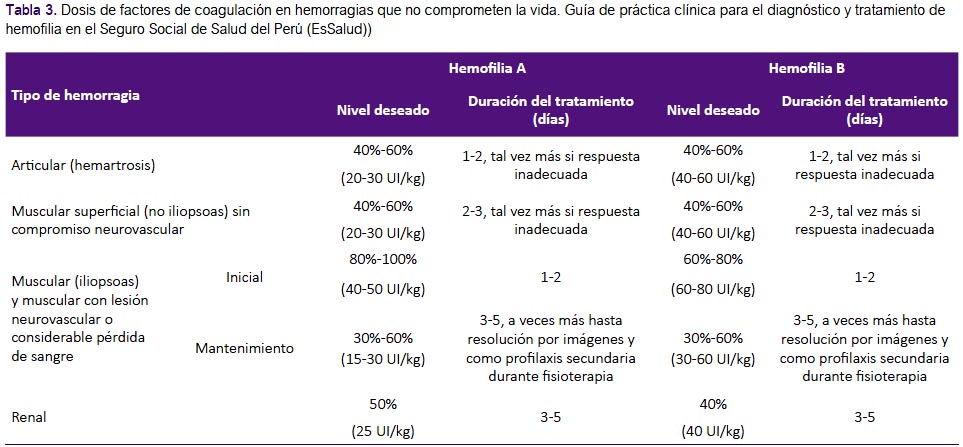
Recomendaciones 12, 13 y 14: La GPC de Chile(2) en base a la guía de la Federación Mundial de Hemofilia(1) recomendó evaluar el estado clínico de cada paciente (incluyendo su estado hemodinámico), y realizar como principal intervención el reemplazo del factor de coagulación deficiente. Una revisión sistemática (12) encontró que el reemplazo del factor de coagulación, en tres condiciones hemorrágicas que no comprometen la vida (hemartrosis, hemorragia muscular y hematuria), tiene efecto sobre la disminución de disfunción y discapacidad a largo plazo, en el paciente. Por ello, se consideró que el reemplazo de factor tiene un balance beneficioso, y con riesgo mínimo para el paciente. Complementariamente, se decidió adoptar las recomendaciones con respecto a la dosis y duración de tratamiento de la GPC de Chile(2). (Tabla 3)
Recomendación 15: Se halló una reciente revisión sistemática Cochrane (13) que evaluó la eficacia del factor VII activado recombinante (rFVIIa) comparado con concentrados derivados de plasma humano (concentrado de FVIII o FIX humano o recombinante a altas dosis; concentrado del complejo protrombínico activado [CCPA]; o concentrado del complejo protrombínico no-activado [CCP]) en el tratamiento de sangrado agudo (principalmente hemartrosis) en personas con hemofilia e inhibidores. Solo dos ensayos clínicos aleatorizados (ECA) (n=69) fueron elegibles para el análisis. Ambos tipos de tratamiento mostraron un efecto hemostático similar en los dos ECA, sin incrementar el riesgo tromboembólico. Debido a que el CCPA tiene una eficacia y seguridad similar a las otras opciones, y que es menos costoso, el GEG-Local decidió proponerlo como primera alternativa para sangrado no vital en pacientes hemofílicos con inhibidores.
Pregunta 6. ¿Cuáles son los tratamientos profilácticos de un paciente hemofílico (A y B) para prevenir daño articular?
La profilaxis en pacientes hemofílicos consiste en reemplazar el factor de coagulación ausente antes que la enfermedad sea activa (primaria) o en un paciente adulto con enfermedad severa para evitar la rápida progresión (secundaria). Esta es una terapia a largo plazo regular y continua. Generalmente, se inicia antes del daño articular, antes de la segunda hemorragia articular clínicamente evidente, o antes de los 3 años de edad (1).
Recomendación 16: La GPC de Chile (2) recomienda el uso de profilaxis primaria en pacientes con hemofilia basado en una RS que halló que el tratamiento profiláctico disminuyó la frecuencia de hemartrosis en comparación con placebo(14) y otra RS que halló que el manejo profiláctico disminuyó el riesgo de todo tipo de sangrado y sangrado vital en comparación al tratamiento a demanda(13).
La RS de Iorio (2011) halló que la terapia profiláctica reducía el riesgo de todo tipo de sangrado (RR: 0,30; IC 95%: 0,12 a 0,76) y de hemartrosis (RR: 0,22; IC 95%: 0,08 a 0,63) en comparación al tratamiento a demanda. Además, esta RS halló que el riesgo de desarrollo de hemartrosis en pacientes con hemofilia A fue menor en pacientes que recibieron profilaxis en comparación a aquellos que recibieron placebo (riesgo atribuible [RA]: -10,73; IC 95%: -16,55 a -4,91)(13). Mientras que la RS de Stobart (2006) halló que el riesgo de sangrado en pacientes con hemofilia A fue menor en aquellos que usaron terapia profiláctica en comparación a aquellos que usaron placebo (RA: -10,80; IC 95%: -16,33 a -5,27) (14). Asimismo, el ECA de Verma del año 2016 halló que el número de episodios de sangrado en pacientes con hemofilia A que recibían tratamiento profiláctico (0,48 ± 0,34) fue menor que el número de episodios en aquellos que recibieron tratamiento a demanda (0,08 ± 0,13) (valor p<0,05)(15). Concordante-mente, los estudios de costo-efectividad identificados concluyeron que el uso de terapia profiláctica sería costo-efectivo en Colombia y en Alemania,(16, 17).
Por ello, el GEG-Local decidió recomendar el uso de terapias de profilaxis. Con respecto a la dosis, se decidió recomendar los protocolos Malmö y Utrecht, según lo propuesto por la Federación Mundial de Hemofilia.(1)
Además, se acotó que en el caso de pacientes en los que se tenga dificultades para el acceso venoso, se puede utilizar el protocolo canadiense de terapia escalonada que ha demostrado ser igual de eficaz que los esquemas antes mencionados(18, 19). Este protocolo establece las siguientes dosis y frecuencias de infusiones de factor VIII:
-
Paso 1: 50 unidades/kg una vez a la semana
-
Paso 2: 30 unidades/kg dos veces por semana
-
Paso 3: 25 unidades/kg en días alternados (puede incrementarse en dosis de 5 unidades/kg si persiste el sangrado)
Si bien diversos estudios han encontrado que esta profilaxis sería costo-efectiva y permitirían ahorrar recursos (16, 17), cabe resaltar que no hay estudios que evalúen la costo-efectividad de la terapia profiláctica en nuestro medio.
Pregunta 7: ¿Cuándo se debe iniciar el tratamiento profiláctico en pacientes hemofílicos?
Recomendación 17: La GPC de Chile (2), en base a la guía de la Federación Mundial de Hemofilia (1) y a una RS Cochrane 2010(13), recomendó iniciar la profilaxis primaria dentro de los tres primeros años de vida y con un máximo de un episodio de hemartrosis. La RS Cochrane 2010 (13) citada resumió seis ECA (n=142) y comparó la profilaxis primaria versus el tratamiento según demanda (factor VIII), encontrando menor riesgo para todo tipo de sangrado (RR: 0,3; IC 95%: 0,12 a 0,76) y para sangrado de articulaciones (RR: 0,22; IC 95%: 0,08 a 0,63) en el grupo de tratamiento profiláctico. También se encontró que una profilaxis iniciada tempranamente previene el desarrollo de inhibidores y ayuda a prevenir las hemorragias con riesgo vital.
Adicionalmente, en base a las recomendaciones de la guía de la Federación Mundial de Hemofilia(1) se establecieron los criterios de inclusión para profilaxis primaria: pacientes con actividad del factor plasmático <2%, edad menor de 3 años, máximo una hemartrosis. (Figura 4)
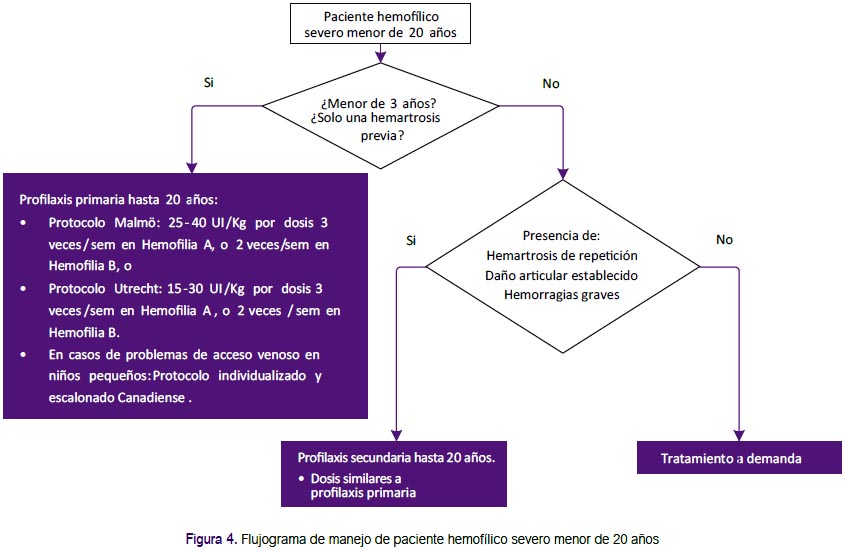
Recomendación 18: Debido a la falta de evidencia que demuestre la mejor edad para descontinuar la profilaxis, el GEG-Local estableció por consenso que la profilaxis primaria se deba mantener por lo menos hasta los 20 años de edad, el cual va acorde a la guía de la Federación Mundial de Hemofilia (1) y posteriormente debe proporcionarse tratamiento a demanda o iniciar profilaxis secundaria según criterios establecidos.
Recomendación 19: Estudios observacionales sugieren que la profilaxis secundaria continua, si bien no revierte el daño articular, puede mejorar el curso de la enfermedad (20). Por ello, se decidió recomendar la realización de profilaxis secundaria a todo paciente en el cual ya existe daño articular y que no entre en el grupo anterior de profilaxis primaria.
Adicionalmente, en base a las recomendaciones de la guía de la Federación Mundial de Hemofilia (1), se establecieron los criterios de inclusión para profilaxis secundaria: Niño con hemofilia severa que no inicio profilaxis primaria por cualquier causa, paciente adulto con hemofilia severa y con eventos hemorrágicos frecuentes (3 o más hemartrosis en la misma articulación en tres meses seguidos) que lo mantienen en reposo continuo y con un alto consumo mensual de factor.
Pregunta 8. ¿Cómo debe ser el manejo de la hemofilia, en niños y adultos en cirugía mayor y menor?
Los pacientes con hemofilia que requieran alguna cirugía mayor o menor deberán recibir un manejo especial. Para esta pregunta, se clasificará a las cirugías en mayores y menores de acuerdo al consenso español (21). (Material suplementario N° 3).
Al respecto, la GPC de Chile (2), recomienda lo siguiente: 1) Procedimientos odontológicos: para limpieza mayor o anestesia de bloqueo para obturaciones o tratamiento de conducto se debe elevar los niveles de factor de un 20 a 30% con una sola dosis, y administrarlo con ácido tranexánico de 30 a 50 mg/Kg/día repartido en tres dosis durante cinco a siete días. 2) Extracciones dentales: elevar los niveles de factor de un 20 a 30% para piezas temporales, 40% para piezas definitivas en una dosis diaria y repetirla a las 24h. Además, recomienda administrarlo con ácido tranexánico de 30 a 50 mg/Kg/día repartido en tres dosis por 10 días, y sellante de fibrina. 3) Cirugía mayor: elevar los niveles de factor de 80 al 100% y luego mantener un factor de 50% en los próximos cuatro días. Mantener los siguientes 10 a 14 días un nivel apropiado en caso de cirugías mayores, y hasta seis semanas en caso de cirugía traumatológica. 4) Cirugía menor: elevar niveles de factor antes de realizar procedimientos diagnósticos invasivos, tales como punciones lumbares, determinación de gases arteriales, broncoscopía con lavado o biopsia y endoscopía digestiva con biopsia.
Recomendación 20: Se llegó a esta recomendación por consenso. El GEG-Local consideró que, debido al riesgo elevado de complicaciones durante un procedimiento quirúrgico, los pacientes con hemofilia deberían ser atendidos en centros de alta complejidad.
Recomendación 21: Se llegó a esta recomendación por consenso. El GEG-Local consideró que debido a que la presencia de inhibidores dirige el manejo de un paciente con hemofilia, sería importante tener esta información antes de realizar el procedimiento quirúrgico. Así, en caso de presentarse una complicación, se podría actuar de forma inmediata.
Recomendación 22: Una RS no encontró diferencias en necesidad de terapia adicional en pacientes sometidos a cirugía menor (1 ECA, OR: 2,37; IC 95%: 0,08 a 66,88) ni para cirugía mayor (1 ECA, OR: 7,5; IC 95%: 0,46 a 122,70) al comparar dosis bajas versus dosis altas de rFVIIa en pacientes con inhibidores (22).
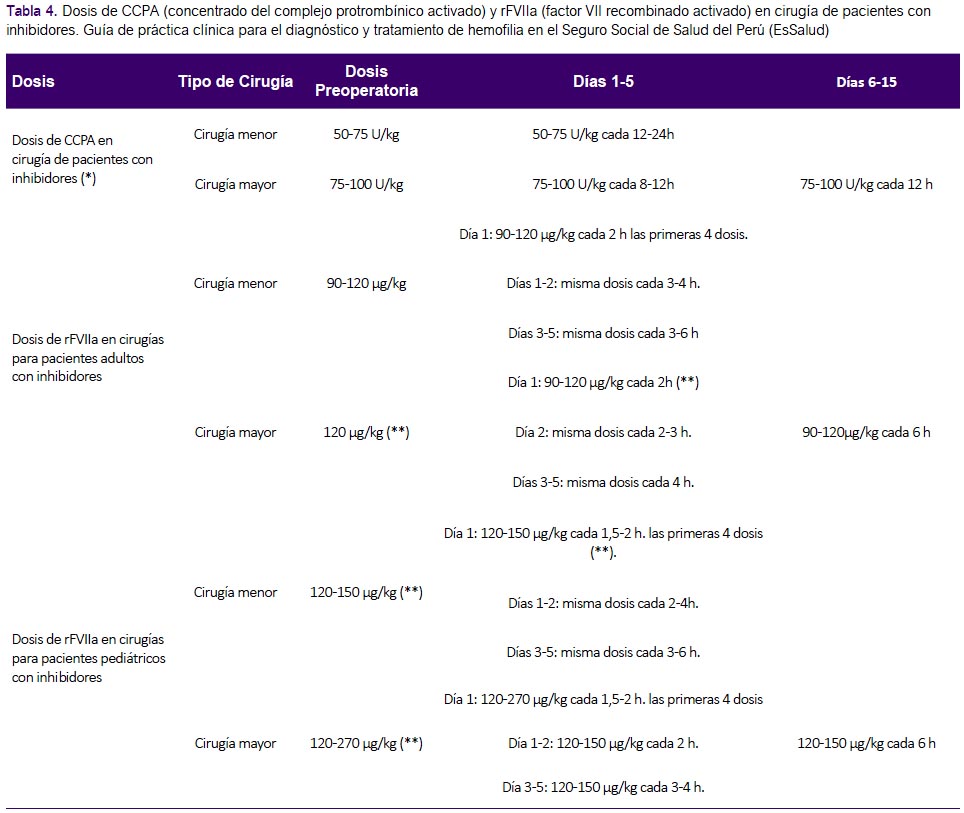
A pesar de esta evidencia no concluyente, se consideró que realizar un manejo perioperatorio tenía potenciales beneficios importantes, y un bajo riesgo; por lo cual se decidió formular una recomendación a favor de este manejo. Las dosis de CCPA y rFVIIa para adultos y niños se adoptaron de GPC de Chile (2). (Tabla 4)
AGRADECIMIENTOS
Agradecemos al Dr. Juan Ramón Navarro Cabrera, presidente de la Sociedad Peruana de Hematología, por la revisión que realizó a la presente guía.
MATERIAL SUPLEMENTARIO
Los materiales suplementarios se encuentran disponibles en formato electrónico en la página web de la revista Anales de la Facultad de Medicina: http://revistasinvestigacion.unmsm.edu.pe/index.php/anales/index.
REFERENCIAS BIBLIOGRÁFICAS
1. The Treatment Guidelines Working Group - World Federation of Hemophilia (WFH). Guidelines for the Management of Hemophilia, second edition. Accedido: Marzo 2016; 2012 Available from: https://www.wfh.org/en/resources/wfh-treatmentguidelines.
2. Ministerio de Salud de Chile. Guía Clínica Hemofilia. Accedido: Marzo 2016; 2013 Available from: http://www.minsal.cl/sites/default/files/Guia_Hemofilia.pdf. [ Links ]
3. Andrews JC, Schünemann HJ, Oxman AD, Pottie K, Meerpohl JJ, Coello PA, et al. GRADE guidelines: 15. Going from evidence to recommendation—determinants of a recommendation's direction and strength. Journal of clinical epidemiology. 2013;66(7):726-35. DOI: 10.1016/j.jclinepi.2013.02.003. [ Links ]
4. Fijnvandraat K, Cnossen MH, Leebeek F, Peters M. Diagnosis and management of haemophilia. Bmj. 2012;344(2707):1-5. DOI: 10.1136/bmj.e2707. [ Links ]
5. Bansal D, Oberoi S, Marwaha R, Singhi SC. Approachto a child with bleeding in the emergency room. The Indian Journal of Pediatrics. 2013;80(5):411-20. DOI: 10.1007/s12098-012-0918-2. [ Links ]
6. Blanchette V, Key N, Ljung L, Manco- Johnson M, Berg H, Srivastava A. Definitions in hemophilia: communication from the SSC of the ISTH. Journal of Thrombosis and Haemostasis. 2014;12(11):1935-9. DOI: 10.1111/jth.12672. [ Links ]
7. Verbruggen H, Novakova I, Wessels J, BoezemanJ, Berg Mvd, Mauser-Bunschoten E. The Nijmegen modification of the Bethesda assay for factor VIII: C inhibitors: improved specificity and reliability. Thromb Haemost. 1995;73(2):247-51. [ Links ]
8. Timmer M, Pisters M, Kleijn P, Bie R, Fischer K, Schutgens R. Differentiating between signs of intra articular joint bleeding and chronic arthropathy in haemophilia: a narrative review of the literature. Haemophilia. 2015;21(3):289-96. DOI:10.1111/hae.12667. [ Links ]
9. Melchiorre D, Linari S, Innocenti M, Biscoglio I, Toigo M, Cerinic M, et al. Ultrasound detects joint damage and bleeding in haemophilic arthropathy: a proposal of a score. Haemophilia. 2011;17(1):112-7. DOI: 10.1111/j.1365-2516.2010.02380.x. [ Links ]
10. Ceponis A, Wong- Sefidan I, Glass C, Drygalski A. Rapid musculoskeletal ultrasound for painful episodes in adult haemophilia patients. Haemophilia. 2013;19(5):790-8. DOI: 10.1111/hae.12175. [ Links ]
11. Bianco RP, Ozelo MC, Villaça PR, Solano MH, Cruz GJ, Murillo CM, et al. Diagnosis and treatment of congenital hemophilia with inhibitors a Latin American perspective. MEDICINA (Buenos Aires). 2008;68(3):227-42. [ Links ]
12. Berntorp E, Astermark J, Baghaei F, Bergqvist D, Holmström M, Ljungberg B, et al. Treatment of haemophilia A and B and von Willebrand’s disease: summary and conclusions of a systematic review as part of a Swedish health- technology assessment. Haemophilia. 2012;18(2):158-65. DOI: 10.1111/j.1365-2516.2011.02723.x.
13. Iorio A, Matino D, D’Amico R, Makris M. Recombinant Factor VII a concentrate versus plasma derived concentrates for the treatment of acute bleeding episodes in people with haemophilia and inhibitors. Cochrane Database Syst Rev.2010;8(8):CD004449. DOI: 10.1002/14651858.CD004449.pub3.
14. Stobart K, Iorio A, Wu JK. Clotting factor concentrates given to prevent bleeding and bleeding-related complications in people with hemophilia A or B. Cochrane Database Syst Rev. 2006;2(2):CD003429. DOI: 10.1002/14651858.CD003429.pub3. [ Links ]
15. Verma S, Dutta T, Mahadevan S, Nalini P, Basu D, Biswal N, et al. A randomized study of very low- dose factor VIII prophylaxis in severe haemophilia–A success story from a resource limited country. Haemophilia. 2016;22(3):342-8. DOI: 10.1111/hae.12838. [ Links ]
16. Molina JO, Duque J, Gutierrez-Ardila M. Cost-Effectiveness Analysis Of Prophylaxis Vs On-Demand Supply Of Factor Ix In Patients Diagnosed With Moderate Hemophilia B In Colombia. Value in Health. 2014;17(3):A228. DOI: 10.1016/j.jval.2014.03.1331. [ Links ]
17. Kip M, den Bosch Mv, Fischer K, Tamminga R, Lepage-Nefkens I. Cost Effectiveness Analysis Evaluating Factor Viii As Primary Prophylaxis Treatmentfor Patients With Severe Haemophilia A In the Netherlands. Value in Health. 2014;17(7):A531. DOI: 10.1016/j.jval.2014.08.1687. [ Links ]
18. Feldman B, Pai M, Rivard G, Israels S, POON MC, Demers C, et al. Tailored prophylaxis in severe hemophilia A: interim results from the first 5 years of the Canadian Hemophilia Primary Prophylaxis Study. Journal of Thrombosis and Haemostasis. 2006;4(6):1228-36. DOI: 10.1111/j.15387836.2006.01953.x. [ Links ]
19. Dodd C, Watts R. A comparison of traditional vs. Canadian tailored prophylaxis dosing of prophylactic factor infusions in children with haemophilia A and B in a single hemophilia treatment center. Haemophilia. 2012;18(4):561-7. DOI: 10.1111/j.13652516.2011.02741.x. [ Links ]
20. Dijk K, Fischer K, Bom J, Scheibel E, Ingerslev J, Berg H. Can long- term prophylaxis for severe haemophilia be stopped in adulthood? Results from Denmark and the Netherlands. British journal of haematology. 2005;130(1):107-12. DOI: 10.1111/j.1365-2141.2005.05546.x. [ Links ]
21. Mingot-Castellano ME, Álvarez- Román MT, López-Fernández MF, Altisent-Roca C, Canaro- Hirnyk MI, Jiménez-Yuste V, et al. Spanish consensus guidelines on prophylaxis with bypassing agents for surgery in patients with haemophilia and inhibitors. European journal of haematology. 2016;96(5):461-74. DOI: 10.1111/ejh.12730. [ Links ]
22. Coppola A, Windyga J, Tufano A, Yeung C, DiMinno MND. Treatment for preventing bleedingin people with haemophilia or other congenital bleeding disorders undergoing surgery. The Cochrane Library. 2015;9(2):CD009961. DOI: 10.1002/14651858.CD009961.pub2fsd. [ Links ]
Conflictos de interés: Ninguno.
Fuentes de financiamiento: Instituto de Evaluación de Tecnologías en Salud e Investigación (IETSI) del Seguro Social de Salud del Perú (EsSalud).
Contribuciones de autoría: Todos los autores participaron en la realización de la guía. AVH y APR se encargaron de las búsquedas sistemáticas y la evaluación de calidad de los estudios para cada pregunta. Todos los autores participaron en la discusión de los estudios encontrados y la formulación de las recomendaciones y puntos de buenas prácticas clínicas.
Correspondencia:
Raúl Timaná-Ruiz
raul.timana@essalud.gob.pe,
rtimanar@gmail.com
Instituto de Evaluación de Tecnologías en Salud e Investigación, EsSalud.
Jirón Domingo Cueto 109, Jesús María, Lima, Perú.
Teléfono: 051-265 6000 anexo: 1953
Recibido: 13 marzo 2018
Aceptado: 4 abril 2018

