










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. vol.79 no.4 Lima oct./dic 2018
http://dx.doi.org/10.15381/anales.v79i4.15637
REPORTE DE CASO
Rara presentación de polineuropatía distal hipoglicémica causada por un insulinoma en el contexto de una neoplasia endocrinamúltiple tipo 1
An unusual presentation of hypoglycemic distal polyneuropathy caused by an insulinoma in the context of multiple endocrine neoplasia 1
Oscar Castillo Sayán1, Cecilia Medina Sánchez1, Roger Verona Rubio2, Enrique Machicado Zuñiga3, German Mendoza Pérez4, Lida Tello Cebreros5
1 Servicio de Endocrinología, Hospital Nacional Arzobispo Loayza. Lima, Perú.
2 Departamento de Patología, Hospital Nacional Arzobispo Loayza. Lima, Perú.
3 Departamento de Cirugía, Hospital Nacional Arzobispo Loayza. Lima, Perú.
4 Departamento de Medicina Nuclear, Instituto Peruano de Energía Nuclear. Lima, Perú.
5 Oficina de Epidemiología, Hospital Nacional Arzobispo Loayza. Lima, Perú.
RESUMEN
La neoplasia endocrina múltiple tipo 1 es una enfermedad de rara presentación. Se caracteriza por el compromiso tumoral neuroendocrino, de paratiroides, hipófisis y enteropancreático. Presentamos el caso de una paciente de 19 años con síntomas de cefalea, convulsiones y debilidad de las cuatro extremidades. Se confirmó la presencia de hipoglicemia 33mg/dL. El estudio elecromiográfico evidenció polineuropatía sensitivo motora en las cuatro extremidades. La resonancia magnética abdominal mostró un tumor en la cola del páncreas que luego de la pancreatectomía se confirmó como insulinoma. La glicemia se normalizó. Además, presentó un macroadenoma hipofisario, hiperparatiroidismo primario y tumor adrenal no funcionante. A los 25 años presentó cefalea intensa y amaurosis de ojo derecho, en la tomografía axial se evidenció tumoración hipofisaria gigante y en estudio de patología se diagnosticó neoplasia maligna condroide (cordoma).
Palabras clave: Neoplasia Endocrina Múltiple tipo 1; Hipoglicemia; Polineuropatía; Insulinoma; Cordoma.
ABSTRACT
Multiple endocrine neoplasia type 1 is a rare disease. It is characterized by the neuroendocrine, parathyroid, pituitary, and enteropancreatic tumor involvement. We present the case of a 19 year old patient with symptoms of headache, convulsions and weakness of the four extremities. The presence of hypoglycemia 33mg/dL was confirmed. The electromyographic study showed motor sensory polyneuropathy in all four extremities. The abdominal magnetic resonance showed a tumor in the tail of the pancreas that after the pancreatectomy was confirmed as insulinoma. The glycemia was normalized. In addition, he presented a pituitary macroadenoma, primary hyperparathyroidism and non-functioning adrenal tumor. At 25 years of age, he presented severe headache and amaurosis of the right eye. Axial tomography showed a giant pituitary tumor and in the study of pathology chondroid malignancy (chordoma) was diagnosed.
Keywords: Multiple Endocrine Neoplasia Type 1; Hypoglycemia; Polyneuropathies; Insulinoma; Chordoma.
La neoplasia endocrina múltiple de tipo 1 es una patología de presentación poco frecuente, con una incidencia anual de 1/30000. Suele comprometer 2 o más glándulas endocrinas tales como paratiroides, páncreas, hipófisis. Es una enfermedad autosómica dominante generada por la presencia de un alelo inactivo del gen MEN 1 (menina). El tejido paratiroideo es comprometido en mayor frecuencia y se encuentra asociado a gastrinoma, insulinoma y prolactinoma1,2. El motivo de presentación del presente caso es la rara asociación entre hipoglicemia crónica causada por insulinoma y polineuropatía periférica. A nivel mundial se han reportado menos de 40 casos que relacionan la presencia de neuropatía por hipoglicemia crónica3,4,5. Además, la paciente presentó una neoplasia maligna de tipo condroide (cordoma), tumor de rara presentación no reportado como componente de MEN tipo 16.
REPORTE DE CASO
Paciente de 19 años, natural de Huaral, Lima. Presentaba un tiempo de enfermedad de un año y cinco meses, caracterizado por presentar cefalea de intensidad moderada, crisis convulsivas tónico clónicas generalizadas y disminución de la fuerza muscular en las 4 extremidades, la cual fue progresiva y ascendente (distal a proximal). Concomitantemente presentó periodos de sensación de frialdad corporal acompañada de temblor generalizado.
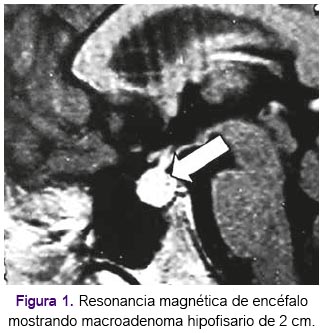
Fue hospitalizada en marzo de 2004 en el Instituto Nacional de Ciencias Neurológicas, con diagnóstico de hipoglicemia (glucosa en 33 mg/dL); durante su hospitalización presentó los siguientes hallazgos clínicos y de imágenes: acné, facie cushingoide, galactorrea, hipotrofia e hipotonía muscular generalizada sin signos meningeos ni sensitivos, macroadenoma hipofisario de 2cm que se extendía a nivel paraselar derecho y que comprime quiasma óptico según resonancia magnética de encéfalo (figura 1), formación hipodensa pararenal izquierda dependiente de glandula suprarenal según tomografía abdominal; y según electromiografía: polineuropatía sensitiva motora con características axonales en las cuatro extremidades a predominio de miembros superiores así como miopatía no inflamatoria de fondo que compromete musculatura proximal. Así, el diagnóstico de la paciente fue de polineuropatía sensitivo motora por hipoglicemia asociado a prolactinoma en relación a una probable neoplasia endocrina múltiple. Durante su hospitalización recibió de tratamiento: dextrosa, cloruro de sodio, cloruro de potasio, dieta completa en 6 servicios diarios, hidrocortisona, prednisona, levotiroxina y cabergolina. Fue dada de alta hospitalaria y referida al Hospital Nacional Arzobispo Loayza (HNAL) para continuación de manejo de paciente.
En abril de 2004 la paciente fue atendida por el Servicio de Endocrinología del HNAL, presentando aún debilidad en las cuatro extremidades, cefalea ocasional, estreñimiento. Se ampliaron sus antecedentes: nacida de parto prematuro con peso al nacer de 1350g, menarquia a los 12 años, regimen catamenial 3 a 4 cada 28 a 45 días, desde los 16 años acné en cara, a los 17 años diagnosticada de tuberculosis ganglionar con tratamiento correspondiente. Al examen clínico: acné en cara, galactorrea en mama derecha, disminución de la fuerza e hipotonía en las cuatro extremidades. Recibió de tratamiento bromocriptina y prednisona.
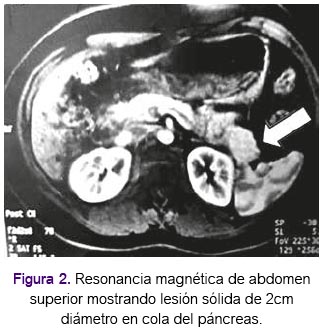
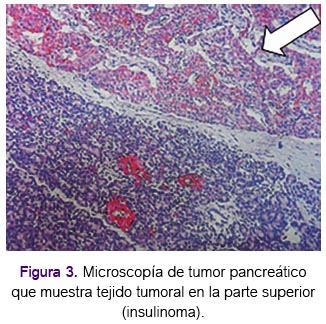
En mayo de 2004 la paciente presentó cuadriparesia, con electomiografía informada como polineuropatía cronica activa (signos de denervación y reinervación) a predominio motor probablemente de tipo axonal. En septiembe de 2004 la resonancia magnética de abdomen superior mostró lesión sólida de 2cm de diámetro dependiente de cola de páncreas, hipercaptadora de sustancia de contraste sugestiva de corresponder a lesión de los islotes, tal como se aprecia en la figura 2. Asimismo, la resonancia mostró lesión nodular de 17mm en glándula suprarenal izquierda. En diciembre de 2004 se le pracitó endoscopía digrestiva alta con hallazgo de tumor submucoso gástrico en cuerpo, gastritis crónica superficial cuya patología mostró gastritis crónica moderada a severa sin displasia ni metaplasia. Con la sospecha clínica y de examenes de un probable insulinoma, se le realizó una pancreatectomía distal cuya anatomía patológica confirmó la presencia de insulinoma. (Figura 3).
En febrero de 2005 la gammagrafía con sestamibi realizada mostró imagen sugestiva de glándula paratiroidea inferior derecha hiperfuncionante con wash out rápido. En noviembre del mismo año la electromiografía de control determinó polineuropatía de tipo axonal en proceso de recuperación.
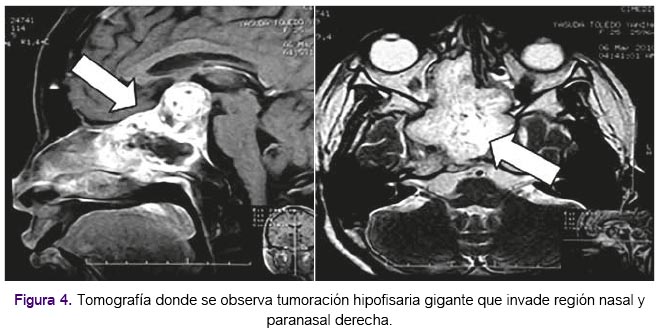
Durante los años 2007 y 2008 la paciente acudió a consulta externa en el HNAL de manera irregular; recibió tratamiento con bromocriptina manteniendo niveles de prolactina, glicemia y calcio dentro de límites normales. En la tabla 1 se muestran los valores de la analítica encontrada en la paciente desde el año 2004 hasta el año 2008. En enero de 2010 acude a la Emergencia del Hospital Nacional Dos de Mayo por presentar cefalea, náuseas, vómitos, amaurosis de ojo derecho y ptosis de párpado derecho, durante 3 semanas de evolución. Fue internada y evaluada por neurocirugía que confirmó tumoración hipofisaria gigante que invade la región nasal derecha tal como se aprecia en la figura 4, según tomografía realizada. Fue intervenida quirúrgicamente por vía transcraneal en marzo de dicho año, encontrándose un macroadenoma con extensión a senos paranasales y fosa nasal derecha, realizándose exéresis parcial del tumor, y un hallazgo patológico de neoplasia maligna con diferenciación condroide tipo cordoma, realizado en el Instituto Nacional de Enfermedades Neoplásicas a donde fue referida para continuar manejo especializado. Según información de los familiares, posteriormente la paciente falleció.

DISCUSIÓN
La neoplasia endocrina múltiple es una patología rara, con incidencia anual de 1/30000, y se define por la presencia de un caso tumoral que implique dos de los órganos siguientes: paratiroides, tejido endocrino enteropancreático e hipófisis anterior. Es una enfermedad autosómica dominante con expresión variable, en la que los individuos afectados heredan un alelo inactivo del gen MEN1 (cromosoma 11q13). Como consecuencia de la inactivación del alelo normal se produce tumorigénesis en tejidos específi cos1,2,7.
El caso de nuestra paciente inició su enfermedad a los 17 años con compromiso del sistema nervioso periférico relacionado con episodios de hipoglicemia crónica evidenciada con datos clínicos, bioquímicos y por valoración de velocidad de conducción nerviosa y electromiográficos. Posterior a la extirpación del insulinoma la glicemia e insulinemia se normalizaron y se observó mejoría del compromiso de la polineuropatía clínica y en el estudio electromiográfico. Es por ello que relacionamos estos cambios con la normalización de la glicemia. En relación a otras causas de polineuropatía, no encontramos datos en la historia clínica acerca de contacto con metales pesados.
Otra posible causa como la deficiencia de vitamina B12, generalmente presenta incremento del volumen corpuscular medio y la hemoglobina corpuscular media de eritrocitos; además de ser frecuente en personas mayores de 30 años8, por lo que fueron descartados como agente causal de polineuropatía.
El compromiso del sistema nervioso periférico causado por la hipoglicemia asociada a insulinoma es un evento poco común. En el mundo se han reportados menos de 40 casos de neuropatía hipoglicémica3, caracterizada por compromiso polineuropático sensitivo-motor, progresiva, simétrica, ascendente, de predominio distal y en miembros superiores3,4. Se plantea que la hipoglicemia causaría un fenómeno de isquemia y reperfusión en el sistema nervioso periférico. Asimismo, algunos investigadores proponen que el daño es morfológico, lo que explicaría la recuperación incompleta después de la pancreatectomía3. Los experimentos en laboratorio apuntan a que la hipoglicemia es el mediador del daño neuronal debido a que altera el transporte axonal rápido4,5, induciendo degeneración de las fibras nerviosas periféricas, particularmente de las localizadas centralmente. El grado de compromiso del sistema nervioso periférico está relacionado con la severidad, duración y frecuencia de los episodios de hipoglicemia. La incidencia anual del insulinoma es de 0,5 a 1 caso por millón de habitantes, 4 a 7% de ellos está asociado a neoplasia endocrina múltiple tipo 1, siendo en este caso de presentación múltiple, y maligna en 25%. La penetrancia a los 40 años es de 10%, y constituye el segundo tumor endocrino enteropancreático más frecuente, después del gastrinoma2,9. Vergés y col. encontraron que la presencia de tumor endocrino enteropancreático como única manifestación inicial de neoplasia endocrina múltiple tipo 1, retarda en promedio 4 años el diagnóstico10. En relación con el manejo, la cirugía resulta curativa en más de 80%, y el procedimiento indicado es la pancreatectomía distal, pues remueve 85% de la glándula11,12.
Se confirmó la presencia de galactorrea clínica, hiperprolactinemia y un macroadenoma hipofisario; la hiperprolactinemia fue controlada con un agonista dopaminérgico. Los tumores pituitarios ocurren en aproximadamente 40% de los casos sintomáticos de neoplasia endocrina múltiple tipo 1 y son más frecuentes en el sexo femenino. Datos del estudio multicéntrico del Groupe d’Etude des Néoplasies Endocriniennes Multiples10,13, reportaron que entre los 136 pacientes portadores de adenomas hipofisarios, estos son la manifestación inicial en 25% de los casos de neoplasia endocrina múltiple tipo 1 esporádico, y en 17% de los casos familiares; 15% de ellos son no funcionantes y aproximadamente 62% secretan prolactina, 9% son secretores de hormona de crecimiento y 4% producen ambas hormonas. Un estudio realizado en niños y adolescentes también encontró una mayor ocurrencia de prolactinomas14.
Los niveles de calcio estuvieron dentro de límites normales o ligeramente incrementados. Siempre con niveles elevados de parathormona, lo cual confirmaba el hiperparatiroidismo primario. Debido a la prioridad del tratamiento del insulinoma y la leve expresión clínica del hiperparatiroidismo no requirió tratamiento. El hiperparatiroidismo primario está presente en 80 a 100% de los casos de neoplasia endocrina múltiple tipo 1, tiene una penetrancia a los 50 años de 100%9. La edad de presentación es en la tercera década de la vida, siendo menor que en los casos no asociados. Entre el 9 a 30% de casos asociados presentan síntomas de hiperparatiroidismo9. El aporte de la gammagrafía con sestamibi es controversial, por lo que su mayor utilidad radicaría en la reintervención quirúrgica, pues en estos casos de hiperplasia sólo se llega a visualizar la glándula que tenga mayor actividad mitocondrial, aquella con mayor avidez por el radiotrazador, lo que a su vez explicaría el fenómeno de wash out rápido15. El momento de la cirugía es controversial, pues una paratiroidectomía temprana predispondría a recurrencia y a mayor dificultad en la reintervención. El procedimiento propuesto es la paratiroidectomía total con autotransplante heterotópico de las paratiroides resecadas2.
El hallazgo en la tomografía abdominal de un nódulo en la glándula adrenal izquierda suele asociarse con neoplasia endocrina múltiple tipo 1. No se evidenció clínica de disfunción adrenal y el cortisol en varias oportunidades estuvo dentro de límites normales. En relación con el compromiso adrenal, la literatura reporta que 30 a 40% de los casos de neoplasia endocrina múltiple tipo 1, se encuentran en forma incidental lesiones adrenales, que pueden presentarse como adenoma, carcinoma o hiperplasia nodular bilateral. Los tumores adrenales no son considerados lesiones principales y usualmente son no funcionantes16.
Reportamos este caso de neoplasia endocrina múltiple tipo 1 por lo inusual de su presentación, con polineuropatía periférica relacionada con hipoglicemia crónica. Finalmente, durante el seguimiento de la paciente, se detectó por estudio de imagen la presencia de un tumor hipofisario con extensión supraselar con compromiso de la región paranasal y nasal derecha. El estudio anatomopatológico determinó el hallazgo de un cordoma maligno, tumor de rara presentación. En este momento surgió la controversia si la paciente presentó clínicamente un macroadenoma productor de prolactina o desde el inicio de su enfermedad presentó el cordoma. Dicha neoplasia en la mayoría de casos inicia su desarrollo en la región esfeno occipital (clivus) y sacra6,17,18, en el caso presentado, la resonancia no evidenció tumoración en esta zona, sino un macroadenoma hipofisario con extensión supraselar que produjo hiperprolactinemia por compresión del tallo pituitario. No existen reportes de lesión inicial de cordoma a nivel hipofisario6,19. Así, no podríamos aseverar que el condroma fue el causante directo de la hiperprolactinemia. De igual manera, no existen reportes de asociación de cordoma a neoplasia endocrina múltiple tipo 16,17-19, dejando abierta la posibilidad que el presente reporte sea el primer caso de dicha asociación.
REFERENCIAS BIBLIOGRÁFICAS
1. Carney JA. Familial multiple endocrine neoplasia: the first 100 years. Am J Surg Pathol. 2005;29(2):254-274. [ Links ]
2. Marx S, Wells SA. Multiple Endocrine Neoplasia. En: Melmed S, Polonsky K, Larsen PR, Kronenberg H. (editores). Williams Textbook of Endocrinology. EE.UU: Elsevier. 2011:1728-1767.
3. Striano S, Striano P, Manganelli F, Boccella P, Bruno R ,Santoro L, et al. Distal hypoglycemic neuropathy. An insulinoma-associated case, misdiaignosed as temporal lobe epilepsy. Neurophysiol Clin. 2003;33(5):223-227. DOI: 10.1016/j.neucli.2003.08.001. [ Links ]
4. Jaspan JB, Wollman RL, Bernstein L, Rubenstein AH. Hypoglycemic peripheral neuropathyin association with insulinoma: Implication ofglucopenia rather than hyperinsulinism. Medicine.1982;61(1):33-44. [ Links ]
5. Heckmann JG, Dietrich W, Hohenberger W, Klein P, Hanke B, Neundörfer B. Hypoglycemic sensorimotor polyneuropathy associated with insulinoma. Muscle Nerve. 2000;23(12):1891-4. [ Links ]
6. Martinez FJ, Conde E, Manjón P, Ricoy JR, Pérez A. Cordoma: Sus variantes y diagnóstico diferencial. Rev Esp Patol.2007;40(3):135-197. DOI: 10.1016/S1699-8855(07)70071-5. [ Links ]
7. Taboada LB, Vera A, Kattah W, López R, Medina VL, González D. Mutación del gen de la menina: desde el hiperparatiroidismo familiar aislado a la neoplasia endocrina múltiple de tipo 1. Rev Colomb Cir. 2011;26(2):118-130. [ Links ]
8. Martinez KM, Cadabal T, Miguens I, García L. Manifestaciones neurológicas por déficit aislado de vitamina B12. Semergen. 2013;39(5): 245-288. DOI: 10.1016/j.semerg.2012.06.006. [ Links ]
9. Kaltsas GA, Besser MG, Grossman AB. The diagnosis and medical management of advanced neuroendocrine tumors. Endocr Rev. 2004;25(3):458-511. DOI: 10.1210/er.2003-0014. [ Links ]
10. Vergès B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G, et al. Pituitary disease in MEN type1 (MEN1): Data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab. 2002;87(2):457-465. DOI: 10.1210/jcem.87.2.8145. [ Links ]
11. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for Multiple Endocrine Neoplasia Type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9):2990-3011. DOI: 10.1210/jc.2012-1230. [ Links ]
12. Yates C, Newey P, Thakker RV. Challengesand controversies in management of pancreatic neuroendocrine tumours with MEN1. Lancet. 2015;3(11):895-905. DOI: https://doi.org/10.1016/S2213-8587(15)00043-1. [ Links ]
13. Goudet P, Bonithon-Kopp C, Murat A, Ruszniewski P, Niccoli P, Ménégaux F, et al. Gender-related differences in MEN 1 lesion occurrence and diagnosis: a cohort study of 734 cases from Grouped´étude des Tumeurs Endocrines. Eur J Endocrinol. 2011;165(1):97-105. DOI: 10.1530/eje-10-0950. [ Links ]
14. Mindermann T, Wilson CB. Pediatric pituitary adenomas. Neurosurgery.1995;36(2):259-68. [ Links ]
15. Nguyen BD. Parathyroid imaging with Tc-99m sestamibi planar and SPECT scintigraphy. Radiographics. 1999;19(3):601-14. DOI: 10.1148/radiographics.19.3.g99ma10601. [ Links ]
16. Barzon L, Pasquali C, Grigoletto C, Pedrazzoli S, Boscaro M, Fallo F. Multiple endocrine neoplasia type1 and adrenal lesions. J Urol. 2001;166(1):24-7. [ Links ]
17. Ferraresi V, Nuzzo C, Zoccali C, Marandino F, Vidiri A, Salducca N, et al. Chordoma: clinical characteristics, management and prognosisof a case series of 25 patients. BMC Cancer.2010;10:22. DOI: 10.1186/1471-2407-10-22. [ Links ]
18. Gutiérrez-Partida CF, Ramirez-Barrios LR, Delgado-Peña B. Cordomas de la bases del craneo. Rev Mex Neuroci. 2014;15(4):207-210. [ Links ]
19. Khawaja A, Venkatraman A, Mirza M. Clival chordoma: case report and review of recent developments in surgical and adjuvants treatments. Pol J Radiol. 2017;82:670-675. DOI: 10.12659/PJR.902008. [ Links ]
Reporte presentado parcialmente en el 41st. Annual Meeting of the European Association for the Studyof Diabetes en septiembre de 2005, Atenas, Grecia
Conflictos de interés: Los autores declaran no tener conflictos de interés.
Fuentes de financiamiento: Autofinanciado.
Correspondencia:
Oscar Alejandro Castillo Sayán
Instituto Nacional de Biología Andina Hospital Nacional Arzobispo Loayza
Av. Alfonso Ugarte 848. Lima 1.
969349149 - oscar.oculm@gmail.com
Recibido: 2 de julio 2018
Aprobado: 6 de noviembre 2018

