










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. vol.79 no.4 Lima oct./dic 2018
http://dx.doi.org/10.15381/anales.v79i4.15639
REPORTE DE CASO
Resistencia adquirida a inhibidores de tirosina kinasa de tercera generación en cáncer de pulmón de células no pequeñas
Resistance acquired to third generation tyrosine kinase inhibitors in non small cell lung cancer
Thanya Runciman Gozzer1, Carlos Carracedo Gonzáles1
1 Unidad de Tumores Torácicos, Clínica Aliada. Lima, Perú.
RESUMEN
La resistencia a inhibidores de tirosina kinasa (TKI) de tercera generación son un evento cada vez más frecuente en la práctica clínica, a pesar de ello, se desconoce aún los mecanismos exactos mediante los cuales las células tumorales desarrollan dicha resistencia permitiendo sus supervivencia. Se describe un caso de resistencia a osimertinib (AZD9291) en cáncer de pulmón de celulas no pequeñas, evidenciado por secuenciamiento genético (NGS) y su posible planteamiento terapéutico.
Palabras clave: Resistencia a medicamentos; Tirosina; Neoplasia Pulmonar.
ABSTRACT
Resistance to third-generation tyrosine kinase inhibitors (TKI) is an increasingly frequent event in clinical practice; however, the exact mechanisms by which tumor cells develop such resistance allowing their survival are unknown. A case of resistance to osimertinib (AZD9291) in non small cell lung cancer, evidenced by next generation sequencing (NGS) and its possible therapeutic approach is described.
Keywords: Drug Resistance; Tyrosine; Lung Neoplasms.
INTRODUCCIÓN
El cáncer de pulmón es una de las principales causas de muerte a nivel mundial y es la tercera causa mas frecuente de mortalidad por cáncer en los varones y la sexta a nivel de ambos sexos en el Perú1. Actualmente, el tratamiento del cáncer de pulmón en el mundo involucra un conocimiento mayor de la biología molecular de este, de manera que al determinar las mutaciones drivers asociadas, se puedan emplear las terapias blanco dirigidas, obteniéndose mejores resultados en la sobrevida global y tiempo libre a la progresión si se compara con la quimioterapia tradicional basada en platino como lo demuestran múltiples estudios2,3,4.
Sin embargo, al tratar las mutaciones iniciales, las células tumorales con el tiempo desarrollarán nuevas mutaciones que les servirán como vías alternativas de proliferación y supervivencia haciendo insostenible continuar con el mismo tratamiento en el caso de comprobarse una progresión sistémica y requiriendo tratamientos de nuevas generaciones que bloqueen estas mutaciones adquiridas4. Los inhibidores de tirosina kinasas (TKI) son fármacos que tienen como función bloquear dichas enzimas que están implicadas en multiples vías de señalización relacionadas con el crecimiento y multiplicación celular las cuales se encuentran alteradas en las células neoplásicas2.
REPORTE DE CASO
Se presenta el caso de una paciente mujer de 45 años de edad, natural y procedente de Lima, que presentó en noviembre de 2013 trastorno visual y pérdida equilibrio por lo cual se le realizó una resonancia magnética nuclear (RMN) cerebral donde se evidenció múltiples formaciones cerebrales y cerebelosas sugerentes de metástasis. En la tomografía de tórax se determinó una lesión de aspecto neoformativo a nivel de región hiliar derecha, ubicada por detrás del bronquio principal derecho, de 43x32 mm, que abarcaba el borde superior del bronquio principal derecho, adenopatía paratraqueal inferior derecha de 17x9mm y presencia de múltiples micronódulos en ambos campos pulmonares. El resto de estudios realizados no evidenciaron alteración. El resultado de la biopsia fue adenocarcinoma pulmonar, gen EGFR mutado con delección en el exon 19, traslocación del gen ALK negativo. El estadiaje clínico realizado fue IVb (T2N1M1).
La paciente recibió radioterapia holocraneal, la cual culminó en diciembre de 2013, e inició tratamiento sistémico con erlotinib (inhibidor de tirosina kinasa: TKI). El control de imágenes de febrero de 2014 mostró disminución en número y tamaño de lesiones cerebrales y una respuesta torácica >80%, se decidió continuar con la misma medicación hasta mayo del 2015, fecha en la cual acude a emergencia por presentar derrame pleural masivo en hemitórax derecho, 17 meses después de haber iniciado terapia con un TKI. En la toracoscopía se evidenció carcinomatosis pleural; se realizó una segunda biopsia, hallándose la mutación T790M positiva en esta nueva biopsia.
La paciente presentó progresión de enfermedad e inició tratamiento con quimioterapia esquema carboplatino + pemetrexed, recibiendo 4 cursos de junio a agosto de 2015 por no contar con terapia target en ese momento. Durante la reevaluación en setiembre del mismo año se catalogó como enfermedad estable. En octubre de 2015 presentó progresión a nivel local por lo que fue incluida en estudio clínico e inició uso expandido con un inhibidor de tirosina kinasa de tercera generación: osimertinib (AZD 9291).
En una nueva evaluación con PET/CT en marzo 2017, se evidenció engrosamiento nodular hipermetabólico pleural derecho, nódulo de 1cm en región hiliar de ese lado. No se mostraron otras alteraciones; así, se consideró que la enfermedad se encontraba estable, continuando con la misma medicación. Finalmente, en julio de 2017, 23 meses después de haber iniciado osimertinib, la RMN de encéfalo evidenció incremento de tamaño de lesión expansiva de tipo secundaria a nivel de la sustancia blanca de 7mm asociado a moderado edema vasogénico, el cual se habia incrementado significativamente comparado con febrero 2017. Además, en la tomografía de tórax se evidenció una imagen pseudonodular, de reciente aparición, de 1,4cm en el lóbulo inferior izquierdo.
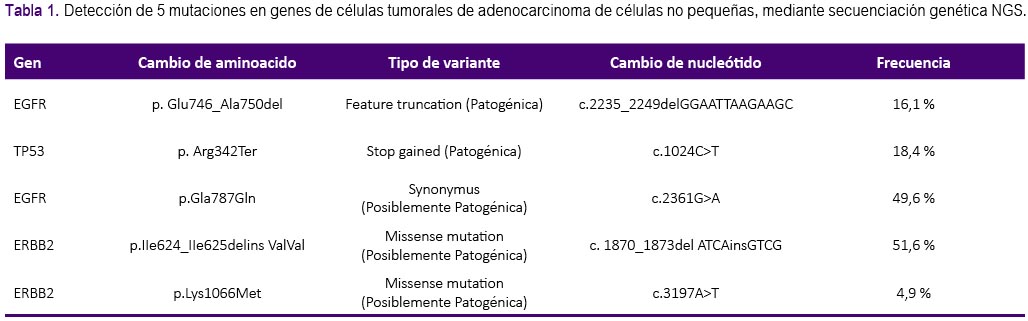
Debido a la progresión pulmonar y cerebral, se decidió realizar una nueva biopsia mediante ecobroncoscopia lineal (EBUS) confirmándose la histología de adenocarcinoma. Dada la evolucion de la enfermedad, se realizó secuenciación genética ultraprofunda (NGS) cuyos resultados fueron mutaciones en los genes: ERBB2 en 51,6%), EGFR Gln787Gln en 49,6%, TP53 en 18,4%, EGFR Glu746_Ala750del en 16,1%, y gen ERBB2 p.Lys1066Met en 4,9%, tal como se aprecia en la tabla 1 donde además se describe el tipo de variante y cambio de nucleótido. No se reportó persistencia de la mutación T790M. También se evaluó la expresión del PD-L1 la cual fue de 0%. Debido a que la paciente se encontraba asintomática y de acuerdo a Niibe y col.5, se cataloga como una oligoprogresión; así, se decidió tratamiento local de las nuevas lesiones con radioterapia y continuación con AZD9291.
DISCUSIÓN
El tratamiento con TKIs como erlotinib, gefitinib y afatinib, en los tumores de pulmón de células no pequeñas (NSCLC, por sus siglas en ingles), aún en pacientes que expresan mutación en dichos receptores, ha demostrado ser superior en el control de la enfermedad metastásica comparado con quimioterapia2,3,4. Sin embargo, aproximadamente el 60% de estos pacientes presentarán durante su evolución diversos mecanismos de mutación, principalmente la mutación T790M5,6. En aquellos pacientes específicamente expuestos a erlotinib, se postula otros mecanismos adicionales de resistencia como la disminución en la expresión de la enzimas relacionadas al glutation y alteración en la vía de factor de transcripción nuclear NRF-2 relacionado con la sensibilidad al erlotinib y cuyo tratamiento con acido etacrinico ha mostrado ser efectivo al resensibilizar dichas células en modelos murinos5.
Como es conocido, la mutación T790M es la causa más frecuente de mutación adquirida, seguida por amplificación de gen MET y transformación en tumor de células pequeñas (SCLC) en un porcentaje muy inferior. Otros mecanismos de resistencia menos frecuentes a TKI de primera o segunda generación, incluyen alteración en la vía de señalización de PI3K, encontrándose cambios en MPK1, HER2 y AXL6. En un estudio donde se analizó sólo población hispana, se observó que la proporción de T790M parece ser similar a lo reportado para la población caucásica; sin embargo, se encuentra una alta proporción de amplificación de EGFR no reportado en otras series6.
Con el objetivo de vencer esta resistencia adquirida se desarrollaron los TKI de tercera generación, como el osimertinib (AZD9291), que inhiben tanto la población T790M wild type así como la mutada, llegando a rangos de respuesta de incluso 60%7. Más allá de un resultado "positivo" o "negativo" para la mutación T790M, lo que determinará la magnitud de la respuesta será la cuantificación de la frecuencia de los alelos, pudiendo ser usado como biomarcador para determinar la sensibilidad del tumor a estas nuevas drogas8.
A pesar de una gran respuesta inicial a los TKI de tercera generación en los pacientes T790M mutados, eventualmente adquirirán mutaciones posteriores a un periodo de aproximadamente 10 meses9, como ocurrió en el caso de nuestra paciente. Además, reportes de biopsias posteriores a la progresión a TKI evidenciaron diferentes patrones de crecimiento en aquellas células tumorales que presentaron la mutación T790M, siendo este mas lento en comparación a las células wild type, confiriendo a la enfermedad un comportamiento mas indolente10. De acuerdo a algunos reportes, la mutación T790M puede no ser detectada en posteriores biopsias7 después de un periodo libre de TKI, como por ejemplo, posterior a tratamiento con quimioterapia11 y pueden ser nuevamente detectada al exponer nuevamente el tumor a un TKI12. Asimismo, la condición de mutación T790M puede variar de acuerdo al lugar de la biopsia -en la misma lesión o en plasma-, y el tiempo en un rango que va 33 a 57% en pacientes con una segunda resistencia adquirida13. Actualmente, la biopsia líquida puede ser utilizada para la determinación y monitoreo de estas mutaciones14.
Oxnar et al. reportó tres tipos de resistencia relacionada al osimertinib, siendo la mas frecuente la mutación C797S, el segundo tipo incluiría tumores T790M positivo sin adquisición de C797S, y el último, pérdidas de T790M con presencia de la mutación EGFR subyacente15. Este ultimo tipo estaría relacionado a nuestra paciente. En la actualidad no existe data suficiente para determinar si es que la mutación T790M se encuentra como parte de una clona mejor controlada e indetectable por exámenes convencionales o si es que realmente ha perdido dicha mutación posterior a la exposición a TKI de tercera generación. Algunos mecanismos distintos han sido relacionados con el rociletinib16. Si consideramos que esta clona de T790M wild type, que se encuentra en un tumor T790M positivo, es resistente también a TKI de primera y segunda generación, debemos pensar que han desarrollado mecanismos de resistencia adicional generando ademas una resistencia cruzada7,17. Sin embargo, otros autores consideran que al desaparecer la mutación T790M estos tumores pueden ser nuevamente expuestos a una terapia con TKI de primera y segunda generación18.
Mecanismos adicionales de resistencia a los TKI de tercera generación, como HER2 y amplificación del MET han sido identificados cuando no se encuentra presente la mutación C797S19. En nuestra paciente, dichas mutaciones se encontrarían en el ERBB2 conservando la mutación del EGFR. La mutación en el Her2 constituye una vía de activación alterna, y su rol ha sido estudiado con mayor frecuencia como causa de resistencia a un TKI de primera generación20. Estudios previos indican que Her2 tiene un rol importante en el crecimiento y sobrevida de las células tumorales21. ErbB2 confiere resistencia a los TKI con acción reversible, reduciendo la tasa de respuesta desde un 25 al 100%22,23. Los tumores con T790M positivo son más propensos a las alteraciones en el HER2, lo cual genera una perdida de la ubiquitinación y subsecuente degradación por el lisosoma24. La amplificación del HER2 es considerado como un mecanismo de resistencia a los TKI en tumores EGFR mutados. Sorprendentemente, EGFR(-), T790M(+), y la amplificación Her2 parecen ser mecanismos mutuamente excluyentes21. No se dispone de información suficiente para determinar si este mecanismo surge como un mecanismo no mediado por la mutación T790M o si se produce debido al uso de fármacos contra dicha mutación25,26. Afatinib, un TKI de segunda generación, tiene actividad contra el EGFR mutado y ademas contra ErbB2, siendo un fármaco atractivo en este escenario27.
Nosotros identificamos al Her2 como potencial mecanismo de resistencia a un TKI de tercera generación en el NSCLC EGFR T790M positivo. Nuestras observaciones sugieren que diferentes clones coexisten en el mismo tumor y serán aquellas T790M wild type las que generan la resistencia a estos nuevos fármacos. Este caso evidencia la importancia de conocer las mutaciones para comprender la heterogeneidad del tumor y escoger la mejor terapia target para tratar de controlar la clona dominante. Se reafirma lo expuesto en la literatura sobre la importancia de las biopsias continuas durante la evolución de la enfermedad, sea esta con biopsia liquida14 o tisular, así como también el momento ideal de su realización.
REFERENCIAS BIBLIOGRÁFICAS
1. Ferlay J, Ervik M, Lam F, Colombet M, Mery L, Piñeros M, Znaor A, Soerjomataram I, Bray F[Internet]. Global Cancer Observatory: CancerToday, 2018. International Agency for Researchon Cancer. World Health Organization. [Fecha de acceso: 15 de agosto 2018]. Disponible en: https://tinyurl.com/y85srtdj. [ Links ]
2. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for Europeanpatients with advanced EGFR mutation-positivenon-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239-46. DOI: 10.1016/S14702045(11)70393-X. [ Links ]
3. Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, et al. Afatinib versus cisplatin-basedchemotherapy for EGFR mutation-positive lungadenocarcinoma (LUX-Lung 3 and LUXLung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16(2):141-51. DOI: 10.1016/S1470-2045(14)71173-8. [ Links ]
4. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DA, Saijo N, et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009; 361:947-957. DOI: 10.1056/NEJMoa0810699. [ Links ]
5. Niibe Y, Hayakawa K. Oligometastases and oligorecurrence: the new era of cancer therapy. Jpn J Clin Oncol. 2010; 40(2): 107–111. DOI: 10.1093/jjco/hyp167. [ Links ]
6. Li H, Stokes W, Chater E, Roy R, de Bruin E, Hu Y, et al. Decreased glutathione biosynthesis contributes to EGFR T790M-driven erlotinib resistance in non-small cell lung cancer. Cell discovery. 2016:2:16031. DOI: 10.1038/celldisc.2016.3. [ Links ]
7. Cardona AF, Arrieta O, Zapata MI, Rojas L, Wills B, Reguart N, et al. Acquired resistance to Erlotinib in EGFR Mutation positive lung adenocarcinoma among Hispanics (CLICaP). Targ Oncol. 2017;12(4):513-523. DOI: 10.1007/s11523-0170497-2. [ Links ]
8. Ichihara E, Lovly CM. Shades of T790M: Intratumor Heterogeneity in EGFR - Mutant Lung Cancer. Cancer Discov. 2015; 5(7):694–6. DOI: 10.1158/2159-8290. [ Links ]
9. Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M, et al. Heterogeneity Underlies the Emergence of EGFR T790Wild-Type Clones Following Treatment of T790M-Positive Cancers with a Third Generation EGFR Inhibitor. Cancer Discov. 2015; 5(7):713–722. DOI: 10.1158/2159-8290. [ Links ]
10. Planchard D, Loriot Y, André F, Gobert A, Auger N,Lacroix L, et al. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Annals of Oncology.2015; 26(10):2073-8. DOI: 10.1093/annonc/mdv319. [ Links ]
11. Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Janne PA, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28(2):357–60. DOI: 10.1200/JCO.2009.24.7049. [ Links ]
12. Kuiper JL, Heideman DA, Thunnissen E, Paul MA, van Wijk AW, Postmus PE, et al. Incidence of T790M mutation in (sequential) rebiopsies in EGFmutated NSCLC-patients. Lung Cancer. 2014;85(1):19-24. DOI: http://dx.doi.org/10.1016/j.lungcan.2014.03.016. [ Links ]
13. Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, et al. Optimization of dosing for EGFR-mutantnon-small cell lung cancer with evolutionary cancer modeling. Sci TranslMed. 2011;3(90):90ra59. DOI: 10.1126/scitranslmed.3002356. [ Links ]
14. Yi-Chen Z, Can P, E-E K, Zhi-Hong C, Jian S, Zhong-Yi, et al. Tracing Spatiotemporal T790M Heterogeneity in Patients with EGFR Mutant Advanced NSCLC after Acquired Resistance to EGFR TKIs. J Thorac Oncol. 2017;12(1):S1249. DOI: https://doi.org/10.1016/j.jtho.2016.11.1762. [ Links ]
15. Esposito A, Criscitiello C, Locatelli M, Milano M, Curigliano G. Liquid biopsies for solid tumors: Understanding tumor heterogeneity and real time monitoring of early resistance to targeted, Pharmacol Ther. 2016;157:120-4. DOI: http://dx.doi.org/10.1016/j.pharmthera.2015.11.007. [ Links ]
16. Thress KS, Paweletz CP, Felip E, Chul B, Stetson D,Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in advanced nonsmall cell lung cancer harboring EGFR T790M. NatMed. 2015; 21(6):560–562. DOI: 10.1038/nm.3854. [ Links ]
17. Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M, et al. Heterogeneity Underlies the Emergence of EGFR T790 Wild-Type Clones Following Treatment of T790M-Positive Cancers with a Third Generation EGFR Inhibitor. Cancer Discov. 2015; 5(7):713–722. DOI: 10.1158/2159-8290. [ Links ]
18. Jong-Mu S, Keunchil P. Can we define the optimal sequence of epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of epidermalgrowth factor receptor-mutant non small cell lung cancer?. Current opinion in oncology. 2017;29(2). DOI: 10.1097/CCO.0000000000000350. [ Links ]
19. Hata A, Katakami N, Kaji R, Fujita S, Imai Y. Does T790M Disappear? Successful Gefitinib Rechallenge After T790M Disappearance in a Patient With EGFR Mutant Non–Small-Cell Lung Cancer. Letters to the Editor. Journal of Thoracic Oncology. 2013;8(3):e27-e29. DOI: https://doi.org/10.1097/JTO.0b013e318282e047. [ Links ]
20. Wang S, Tsui ST, Liu C, Song Y, Liu D. EGFR C797S mutation mediates resistance to thirdgeneration inhibitors in T790M positive non-small cell lung cancer. J Hematol Oncol. 2016; 9(1):59. DOI: 10.1186/s13045-016-0290-1. [ Links ]
21. Kim TM, Song A, Kim DW, Kim S, Ahn YO, Keam B, et al. Mechanisms of Acquired Resistance to AZD9291A Mutation-Selective, Irreversible EGFR Inhibitor. J Thorac Oncol. 2015;10(12):1736-44. DOI: 10.1097/JTO.0000000000000688. [ Links ]
22. Takezawa K, Pirazzoli V, Arcilla M, Nebhan C, Song X, de Stanchina E, et al. HER2 Amplification: A Potential Mechanism of Acquired Resistance to EGFR Inhibition in EGFR -Mutant Lung CancersThat Lack the Second-Site EGFR T790M Mutation. Cancer Discovery. 2012; 2(10): 2(10):1-12. DOI: 10.1158/2159-8290. [ Links ]
23. Landi L, Cappuzzo F. HER2 and lung cancer. Expert Rev Anticancer Ther. 2013;13(10):1219-28. DOI: 10.1586/14737140.2013.846830. [ Links ]
24. Weickhardt AJ, Scheier B, Burke JM, Gan G, Lu X, Bunn PA, et al. Local ablative therapy of oligo progressive disease prolongs disease control by tyrosine kinase inhibitors in oncogeneaddicted non-small-cell lung cancer. J ThoracOncol. 2012;7(12):1807-1814. DOI: 10.1097/JTO.0b013e3182745948. [ Links ]
25. Shtiegman K, Kochupurakkal BS, Zwang Y, Pines G, Starr A, Vexler A, et al. Defective ubiquitinylation of EGFR mutants of lung cancer confers prolonged signaling. Oncogene. 2007; 26(49):6968-78. DOI: 10.1038/sj.onc.1210503. [ Links ]
26. Suda K, Rivard CJ, Mitsudomi T, Hirsch FR. Overcoming resistance to EGFR tyrosine kinase inhibitors in lung cancer, focusing on non-T790M mechanisms. Expert Rev of Anticancer Ther. 2017;17(9):779-786. DOI: 10.1080/14737140.2017.1355243. [ Links ]
27. Marquez-Medina D, Popat S. Afatinib: a secondgeneration EGF receptor and ErbB tyrosine kinase inhibitor for the treatment of advanced non-smallcell lung cancer. Future Oncol. 2015;11(18):2525-40. DOI: 10.2217/fon.15.183. [ Links ]
Conflictos de interés: Los autores declaran no tener conflictos de interés.
Fuentes de financiamiento: Autofinanciado.
Correspondencia:
Thanya Runciman Gozzer
targo18@gmail.com
Recibido: 16 de septiembre 2018
Aprobado: 16 de noviembre 2018

