











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El angioma de células litorales (ACL) es un tumor vascular de bazo, poco frecuente, descrito por primera vez por Falk et al. en 1991 1, pertenece a un grupo de neoplasias entre las que se encuentran también el hemangioendotelioma de células litorales y el angiosarcoma de células litorales 2. Este tumor deriva de las células que rodean los senos de la pulpa roja del bazo, células litorales, encargadas de fagocitar los eritrocitos envejecidos 3. La prevalencia de esta enfermedad es desconocida debido a que es una neoplasia de rara presentación, tampoco se ha descrito predilección de presentación según grupo etario ni sexo 4.
A nivel histológico se caracteriza por tener estructuras usualmente bien delimitadas, no encapsuladas 4,5, con zonas vasculares quísticas tapizadas por células de hábito histocitario/macrofágico que se desprenden hacia la luz, sin atipia o asociación de proliferación linfática 3. A nivel inmunohistoquímico expresan marcadores endoteliales como el CD31, Factor VIII, y marcadores histiocíticos como el CD68. A diferencia de las células litorales normales, en el ACL no se expresa CD8, el cual es un marcador que expresa la conservación de la arquitectura de la pulpa roja del bazo 6.
Las células litorales proliferan en circunstancias de estimulación antigénica, por lo que determinados estudios han demostrado cierta asociación entre el ACL con algunas enfermedades malignas e inmunológicas; sin embargo, en la mayoría de los casos se presenta como un hallazgo incidental, usualmente asintomático, y en los casos que se presentan síntomas estos son muy inespecíficos como masa abdominal, sensación de distensión abdominal, anemia o trombocitopenia 4,7.
Se realizó una revisión de la literatura en las bases de datos PubMed (MedLine) y LILACS, utilizando una combinación de términos MeSH (Medical Subject Heading) como: ("Littoral cell angioma of the spleen "[Supplementary Concept] OR "Littoral cell angioma of the spleen" [All Fields]]), ("Littoral cell angioma of the spleen" [Supplementary Concept]) AND "Tomography, X-Ray Computed"[Mesh] desde el año 2013 hasta la actualidad. Luego de la búsqueda no hallamos otro caso similar reportado en nuestro país por lo que consideramos que el nuestro sería el primer caso publicado de ACL en Perú.
REPORTE DE CASO
Varón de 62 años con antecedente de glaucoma desde los 37 años, fue visto en un hospital de Lima-Perú, el año 2013, con un tiempo de enfermedad de cinco años, presentando una tumoración en hipocondrio izquierdo, de crecimiento progresivo, asociado a dolor, sensación de llenura precoz y pérdida de peso 5kg tres meses antes de la consulta. Al examen físico, se apreció una masa palpable que abarcaba hipocondrio, flanco izquierdo y epigastrio. Los estudios auxiliares solicitados evidenciaron anemia severa (Hb 5,70 gr/dL), trombocitopenia, hipoalbuminemia e hipoglobulinemia. Se realizó una ecografía abdominal que determinó bazo y riñón parcialmente definibles debido a presencia de masa mal delimitada de 16x13cm ubicada en cuadrante superior izquierdo; se solicitó una tomografía computada (TC) con contraste la cual mostró una masa heterogénea, dependiente de bazo que desplazaba (sin infiltrar) estructuras adyacentes, con áreas extensas de calcificación.
Se decidió realizar una laparotomía exploratoria, evidenciándose tumoración esplénica que abarcaba toda su extensión siendo catalogado como irresecable, realizándose una punción-aspiración (PAAF) de la lesión, cuyo reporte patológico describió células neoplásicas de aspecto redondo, no se realizó estudio de inmunohistoquímica debido a la cantidad de la muestra. El paciente acudió al servicio de oncología médica, donde basándose en las imágenes y la histología, se consideró que presentaba un sarcoma por lo que se le pautaron cuatro ciclos de quimioterapia con esquema doxorrubicina + ifosfamida de abril a julio del 2013 sin respuesta y 25 sesiones de radioterapia de agosto a setiembre del 2013, sin respuesta favorable.
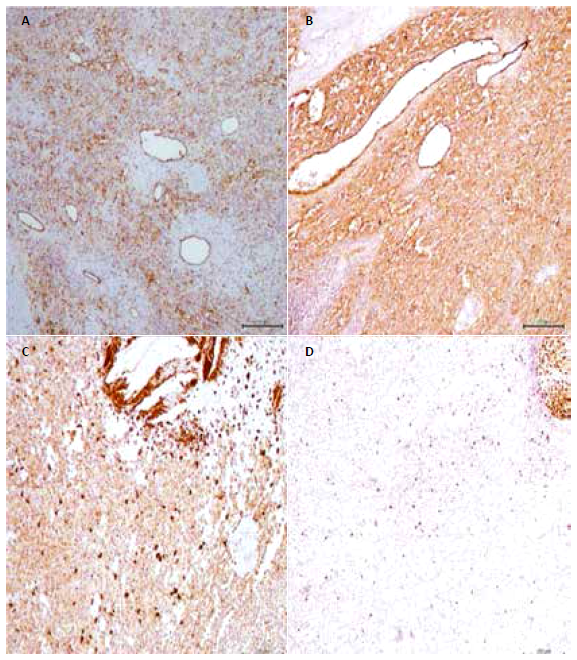
Se reevaluó el caso el año 2014, decidiendo reintervenir quirúrgicamente. En la segunda cirugía se apreció macroscópicamente una masa blanquecina que infiltraba retroperitoneo de consistencia calcificada, ubicada en el polo inferior del bazo, el cual se encontraba macroscopicamente hipotrófico en su parte superior. Se resecó la masa tumoral en bloque y se envió a patología, quienes describieron un bazo de 1800 mg de 22x17x12cm, color grisáceo, con zonas mixoides, hemorrágicas y necróticas, y 30% de áreas calcificadas (Figura 1), de aspecto multinodular con trayectos bien definidos que afectaban casi todo el espécimen. Al examen microscópico se apreciaron canales vasculares con células endoteliales altas y otras de aspecto histiocitario (Figura 2a), con proyecciones papilares (Figura 2b). Las células mostraron hemofagocitosis, hemosiderina luminal (Figura 2c), cristales de colesterol con reacción xantomatosa y calcificación distrófica (Figura 2d). El examen inmunohistoquímico reveló que las células fueron positivas para la línea endotelial CD31, Factor VIII, marcadores histiocíticos CD68, y ki67 negativo (Figura 3). Por todo lo anterior se catalogó al tumor como un angioma de células litorales esplénico. El paciente luego de la cirugía inició controles periódicos sin evidenciar recidiva local o a distancia, hasta la fecha del presente reporte.
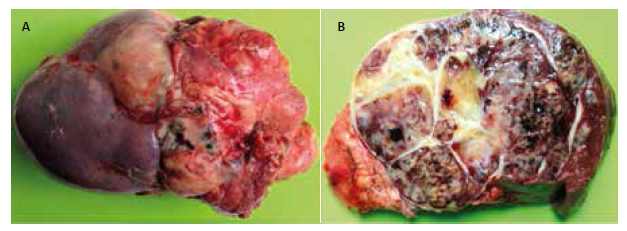
Figura 1. Macroscopía post- esplenectomía que muestra zona de superficie irregular con características quísticas (A). Bazo de 1800 mg de 22x17x12cm, con zonas mixoides, hemorrágicas y necróticas, y 30% de áreas calcificadas (B).
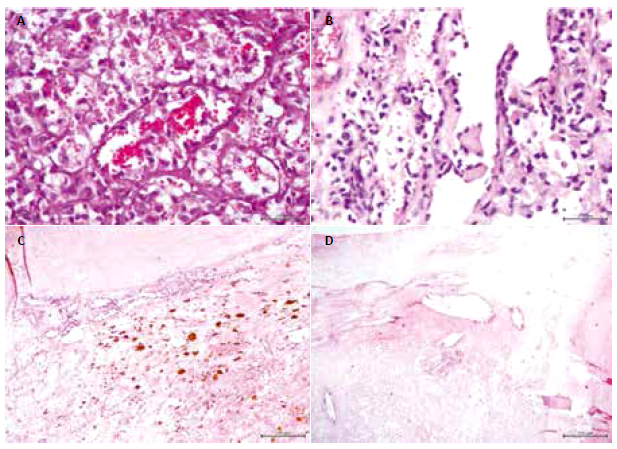
Figura 2. (A) Tinción hematoxilina eosina (H&E), 100x: canales vasculares con células endoteliales altas, otras de aspecto histiocitario. (B) H&E, 100x: vasos con endotelio alto y proyecciones papilares. (C) H&E, 40x: hemofagocitosis. (D) H&E, 20x: cristales de colesterol y reacción xantomatosa.
DISCUSIÓN
El ACL no tiene una presentación etaria específica, las edades fluctúan entre la tercera y octava década de vida y tampoco muestra predominancia de acuerdo a sexo masculino o femenino 4. Generalmente es asintomática; sin embargo, en los pocos casos descritos se presentaron síntomas generales como distensión y dolor abdominal, fatiga, disminución de peso, fiebre y sensación de llenura precoz (4,8) . El paciente del presente reporte presentó dolor abdominal, llenura precoz y disminución de peso.
El ACL puede acompañarse de otras neoplasias hematológicas como leucemia linfocítica crónica y linfoma maligno, así como neoplasias viscerales tales como colorrectales, renales, pancreáticas, meningiomas 4 e inclusive paragangliomas 9. De igual manera en enfermedades autoinmunes como enfermedad inflamatoria intestinal y púrpura trombocitopénica autoinmune 10 y algunos síndromes metabólicos como la enfermedad de Gaucher 9.
Respecto a los parámetros de laboratorio, el ACL se puede presentar asociado a anemia 3, elevación de las transaminasas y trombocitopenia, como en el caso reportado por Jasani et al. en el cual un niño de 2 años presentó trombocitopenia crónica que se resolvió posterior a la esplenectomía, en cuyo estudio anatomopatológico se concluyó el diagnóstico de ACL11. En nuestro caso el paciente presentó anemia, trombocitopenia e hipoproteinemia.
Los estudios de imágenes no son específicos, siendo descritos en las tomografías como lesiones hipodensas, existiendo dos formas de presentación: múltiples nódulos en todo el bazo (la más común) o como un nódulo solitario, motivo por el cual para constatar el diagnóstico siempre es necesaria la realización de una biopsia con aguja fina 4.
Con respecto a los estudios inmunohistoquímicos, la mayoría de casos reportados expresó el marcador CD68, Factor VIII, CD8 y CD34; mientras que, una menor cantidad expresó lisozima y CD31. Peckova et al. revisó 25 casos de ACL y encontró que los marcadores que tuvieron mayor intensidad exclusivamente para las células litorales en el ACL fueron Claudin-5 y VEGFR-2, mientras que WT-1 se expresó fuertemente en el citoplasma de células esplénicas que no tenían ninguna alteración 4. La evaluación inmunohistoquímica en nuestro caso resultó positiva para CD31, Factor VIII, CD68; y fue negativa para Ki67, demostrando así su estirpe de neoplasia benigna con un bajo porcentaje de crecimiento. El marcador Factor VIII y CD31 demuestra la diferenciación endotelial mientras que el marcador CD68, lisozima y CD 21 demostraron la diferenciación histiocítica. Para el diagnóstico de ACL es fundamental demostrar ambas diferenciaciones y nuestro paciente las presentó.
El manejo de este tipo de tumores es quirúrgico y se fundamenta en la esplenectomía, debido a que la evolución de los ACL suele tener un curso benigno. Cai et al. realizó un estudio en el comparó a dos grupos, el grupo 1 se sometió a esplenectomía laparoscópica (13 casos) y el grupo 2 a esplenectomía abierta (14 casos), se evidenció que los pacientes que se sometieron a esplenectomía laparoscópica tuvieron menor tiempo operatorio (128+/-37 min vs 177+/-25 min), menor sangrado (62+/-48 ml vs 138+/-64 ml) y menos complicaciones que la esplenectomía abierta; sin embargo, un caso de esplenectomía laparoscópica requirió ser convertido a esplenectomía abierta12. Li et al. reportaron el caso de una paciente de 32 años que presentaba lesiones múltiples en el bazo, y fue sometida a esplenectomía laparoscópica la cual resultó sin complicaciones 13. Por su parte, Takayoshi et al. reportaron un caso de ACL con afectación metastásica a hígado y confirmación inmunohistoquímica, se trató de una paciente de sexo femenino que inicialmente fue tratada con prednisolona y ciclosporina, con pobre respuesta por lo que recibió posteriormente quimioterapia con etopósido, paclitaxel y vincristina, logrando respuesta parcial a nivel de las lesiones hepáticas de acuerdo a PET-TC (disminución de SUV) 14; sin embargo, este tipo de opciones terapéuticas constituyen una excepción a la regla de acuerdo a los reportes revisados, reservados solamente para enfermedad metastásica.
El paciente que presentamos, luego de la esplenectomía, se encuentra en observación hasta la actualidad. Cabe señalar que debido a un diagnóstico inadecuado, fue catalogado inicialmente como un sarcoma, motivo por el cual recibió quimioterapia y radioterapia secuencial sin obtener respuesta, lo que evidencia la escasa actividad de este tipo de terapias en casos de ACL esplénico; aunque se debería considerar que nuestro paciente fue tratado con quimioterapia para sarcoma y no como ACL metastásico.

