











 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Las enfermedades por priones son una familia de trastornos degenerativos letales causados por la acumulación de partículas transmisibles que contienen una isoforma anómala de la proteína priónica (PRPN) 1. La enfermedad de Creutzfeldt-Jakob (ECJ) es la enfermedad por priones más común en humanos y se clasifica en forma esporádica (esECJ), familiar (fECJ), iatrogénica (iECJ) y variante (vECJ) 1. La incidencia anual reportada es de 1 caso por un millón de habitantes 2. La forma esporádica es la variante más común, presenta mal pronóstico y el 90% de pacientes fallecen dentro del primer año del inicio de síntomas debido a infección o falla cardiorrespiratoria 2. En el Perú, 11 casos, incluyendo seis definitivos y cinco casos probables, han sido reportados en la literatura entre 1998 y el 2013 3.
Presentamos un caso de ECJ probable, con un tiempo de enfermedad de 25 meses, proveniente de una institución privada en Lima, Perú. Nuestro objetivo es describir indicadores clínicos e imagenológicos que traten de explicar dicha sobrevida prolongada.
REPORTE DE CASO
Paciente varón de 66 años, peruano, con una historia de dos meses de pérdida de memoria progresiva, apraxia motora y cambios del temperamento. Visitó a un neurólogo y tras realizar estudios por imágenes donde no se evidenciaron anormalidades, el médico atribuyó que estos síntomas eran secundarios a estrés. Luego de dos meses, los familiares del paciente percibieron el agravamiento de los síntomas previos, junto con la aparición de dificultad para articular palabras y repuestas cortas y simples frente a preguntas. El paciente desarrolló progresivamente una hemiparesia espástica del hemicuerpo derecho y dificultad en la deglución, por lo que fue hospitalizado en Roma, Italia. Allí, fue manejado con ventilación mecánica (VM) debido a una insuficiencia respiratoria y tratado con antibióticos para una neumonía asociada a VM. Un mes más tarde, tras estar estabilizado, la familia decidió transferir al paciente a nuestra institución en Perú: Clínica Angloamericana.
Al examen neurológico inicial, la escala de coma de Glasgow fue de 11, el paciente presentaba hemiparesia del hemicuerpo derecho y pupilas isocóricas, sin otros hallazgos significativos.
Adicionalmente, desarrolló una convulsión mioclónica con desviación ocular a la derecha y fue transferido a la unidad de cuidados intensivos (UCI) para manejo de una insuficiencia respiratoria con VM. Los diagnósticos neurológicos preliminares consideraron una encefalopatía viral, autoinmune o esponpngorme.
El estudio del líquido cefalorraquídeo (LCR) fue positivo para la proteína 14- 3-3, sin otros hallazgos relevantes. La posibilidad de una encefalopatía viral o autoinmune se alejó luego de obtener resultados negativos para anti-VGKC, anti-NMDA, anticuerpos antitiroides y serología negativa para el virus de herpes simple, citomegalovirus, virus de Epstein-Barr, virus JC, sífilis, virus linfotrópico de células T humano y hepatitis. El electroencefalograma (EEG) evidenció actividad cerebral inconstante y compromiso encefálico difuso sin actividad paroxística. Además, se realizó una resonancia magnética (RM) cerebral en donde se encontraron hiperintensidades a nivel de ganglios basales, hallazgo característico de ECJ (Figura 1).
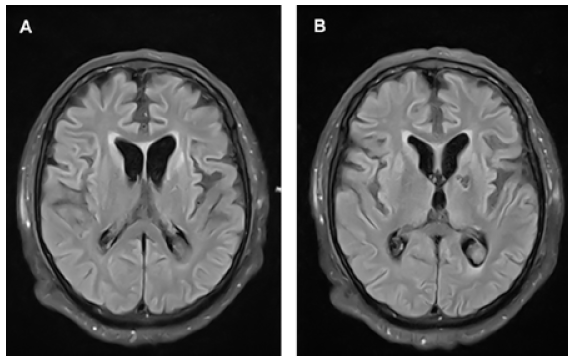
Figura 1 RM en la admisión. (A y B) La secuencia FLAIR muestra hiperintensidades en la cabeza de ambos núcleos caudados, a nivel de las cortezas insulares y en la superficie medial y lateral de la corteza frontal. (B) Se observa además la secuela de un infarto lacunar en la rodilla de la cápsula interna izquierda.
Una semana después, se repitió un nuevo EEG el cual mostró ondas agudas generalizadas, prominentes en las regiones bifrontales y centrales con lateralizaciones a la izquierda, sobre un fondo de ondas delta y teta de bajo voltaje. Una segunda RM cerebral reveló atrofia cortical e hiperintensidades (Figura 2). Aquel día el paciente fue transferido de la UCI al piso de hospitalización.
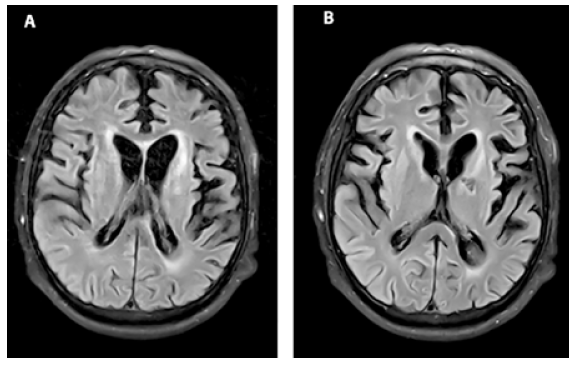
Figura 2 RM 30 días luego de la admisión. (A) La secuencia FLAIR evidencia una dilatación compensatoria del sistema ventricular como resultado de la atrofia cortical moderada del parénquima cerebral. (B) Hiperintensidades multifocales en las cortezas insulares, frontales y occipitales.
De acuerdo a las características clínicas, la proteína 14-3-3 positiva en LCR y los hallazgos en la RM, el paciente cumplía con los criterios para el diagnóstico probable de la variante esporádica de la ECJ 4. Nueve meses luego del alta, durante una hospitalización para el manejo de una neumonía aspirativa se le realizó una nueva RM control donde se evidenciaron cambios degenerativos importantes (Figura 3). Tras la última hospitalización, el paciente permaneció en el mismo es tado neurológico hasta su fallecimiento en casa 25 meses desde el inicio de los síntomas.
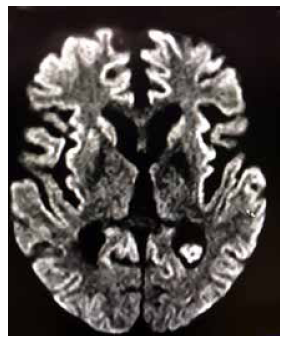
Figura 3 RM 9 meses luego del alta. La secuencia FLAIR muestra cambios degenerativos severos caracterizados por una marcada reducción del volumen cortico-subcortical, dilatación simétrica y global de las cavidades ventriculares (hidrocéfalo ex vacuo) y marcadas hiperintensidades en las cortezas frontales y parietales de forma bilateral, y en la cara medial de los lóbulos occipitales, también bilateralmente.
DISCUSIÓN
La forma esporádica de la ECJ representa aproximadamente el 85% del total de casos de las enfermedades por priones en humanos y la sobrevida promedio es de 4-6 meses 5,6. El paciente presentado con esECJ fue diagnosticado basado en los criterios diagnósticos de la Unidad de Vigilancia e Investigación Nacional de ECJ de Inglaterra 4, y presentó un tiempo de enfermedad total (hasta la muerte) de 25 meses. El rápido reconocimiento de los factores que influyen en la sobrevivencia puede ayudar en la toma de decisiones respecto al manejo del paciente y en la consejería familiar.
La mayoría de casos con ECJ con sobrevida prolongada pertenecen a la población japonesa y existen dos explicaciones posibles: el mayor acceso a tratamientos intensivos de soporte vital que prolongan la vida, y a la mayor prevalencia de la variante panencefalopática de la ECJ (ECJPE), que tiene un tiempo de enfermedad promedio de 22 meses 7,8. Además, la implementación de alimentación por sonda ha sido descrita como un factor esencial que contribuye a sobrevida prolongada en pacientes con esECJ en Japón 9. En el presente caso, dado que la alimentación por sonda y VM estuvieron implicadas, el tiempo de sobrevida pudo haberse prolongado artificialmente, especialmente luego de que el paciente alcanzó el estado de mutismo acinético.
En la forma esECJ, el tiempo promedio entre el inicio de enfermedad y el primer episodio de mioclonías, y hasta alcanzar el estado demutismo acinético es de dos meses y tres meses, respectivamente 10. En este caso, dichos tiempos fueron de cinco meses y siete meses. Además, la duración de la enfermedad se ha asociado con el tiempo desde el inicio de la enfermedad hasta la primera mioclonía y el tiempo hasta alcanzar estado de mutismo acinético 9. Por lo tanto, la progresión clínica relativamente lenta de nuestro caso podría corresponder con un tiempo de enfermedad prolongado, considerando que un inicio agudo y progresión rápida se observan en pacientes con menor tiempo de sobrevida 11,12.
Nuestro paciente presentó un curso de dos estadios: el primer estadio se caracterizó por un deterioro rápido inicial que progresó a mutismo acinético, y el segundo estadio mostró la persistencia del mutismo acinético por 18 meses. Un curso similar de dos estadios se ha descrito en la variante ECJPE 10. Sin embargo esta variante anatomopatológica no ha sido reportada previamente en poblaciones latinoamericanas, probablemente debido a la falta de estudios en autopsia en estos países.
En la etapa temprana de la enfermedad, trastornos de la memoria y cambios de humor llevaron a nuestro paciente a consultar a un médico, donde se le realizó una RM sin anormalidades sugestivas de EJC. En esta fase, la mayoría de pacientes evidencian las imágenes características; sin embargo, la ausencia de hiperintensidades en la secuencia FLAIR o difusión ya ha sido reportada 13,14. Además, los pacientes con imágenes típicas de ECJ podrían presentar un tiempo menor de sobrevida al compararse con aquellos que presentan imágenes atípicas en la RM 11.
Se ha reportado que el compromiso de la sustancia gris se asocia con un tiempo de enfermedad breve y deterioro neurológico rápido 11,15. Luego de cinco meses de enfermedad, nuestro paciente presentó compromiso de ganglios basales en la RM. El tiempo entre el inicio de enfermedad y la primera evidencia de hiperintensidad cortical tuvo una diferencia de 1,6 meses más tarde comparado con el tiempo promedio de la determinación de estos hallazgos 10, lo cual sugiere que el compromiso tardío cortical y de ganglios basales en nuestro caso podría asociarse con la evolución prolongada de la enfermedad. Sin embargo, es relevante considerar que nuestro paciente no presentó imágenes en la secuencia difusión, que es considerada superior a FLAIR en la detección de hiperintensidades corticales 13. Por lo tanto, esa podría ser una limitación en el diagnóstico precoz de ECJ en nuestro caso.
En el Perú, una serie de 11 pacientes fue publicada por Torres-Ramirez y col. 3. Se reportó un tiempo de enfermedad promedio de 8,6 y 9 meses para casos definitivos y probables, respectivamente. Además, uno de los casos probables, un varón de 55 años, presentó un tiempo de enfermedad de 18 meses. De esta forma, nuestro paciente es el caso de esECJ con diagnóstico probable con mayor tiempo de sobrevida reportado en nuestro país. Por otro lado, la ECJ no es parte del sistema nacional de vigilancia epidemiológica del Perú. Además, esta enfermedad es considerada rara en nuestro país, probablemente por limitaciones en su diagnóstico, como los altos costos de la proteína 14-3-3 en LCR y la no disponibilidad de estudios en autopsia en ciertas instituciones para la confirmación diagnóstica. En el presente caso, no se pudo realizar el estudio histológico para alcanzar el diagnóstico definitivo.
La ECJ es una enfermedad rara y poco reportada en el Perú. El tiempo desde el inicio de enfermedad hasta presentar mioclonías y hasta alcanzar el mutismo acinético, y la implementación de terapias de soporte fueron factores que contribuyeron a la larga sobrevida en este caso de esECJ probable. Considerando las limitaciones de la secuencia FLAIR, la ausencia de hallazgos precoces en imágenes, y el compromiso tardío de corteza y de ganglios basales, también podrían explicar el tiempo de enfermedad de nuestro paciente.

