











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El linfoma angioinmunoblástico de células T (LAIT) fue descrito por primera vez por Frizzera en 1974 como una linfoadenopatía angioinmunoblástica con disproteinemia 1. Ahora se sabe que se trata de un linfoma no Hodgkin de células T poco frecuente y que representa el 15 al 20 % de los linfomas T periféricos 2,3. Sus manifestaciones clínicas son variables e inespecíficas y se basan en síntomas y signos generales, sin embargo, puede manifestarse como cuadros paraneoplásicos imitando enfermedades autoinmunes 2,4.
El diagnóstico se basa en la inmunohistoquímica y el pronóstico en general es pobre, reportándose una sobrevida a los 5 años de 30 % 4. Por lo tanto, un diagnóstico precoz ayuda a iniciar el tratamiento oportuno, lo cual mejora el pronóstico de los pacientes que lo padecen. Este reporte tiene como objetivo hacer un análisis de la presentación del LAIT como imitador de enfermedades autoinmunes; por lo que, presentamos un caso con diagnóstico inicial clínico y laboratorial de lupus eritematoso sistémico, diagnóstico que fue replanteado al no encontrar mejoría con el tratamiento y presentar, además, la aparición de otros signos clínicos.
REPORTE DE CASO
Mujer de 36 años natural de Lima, sin antecedentes de importancia, acude con enfermedad de 3 años caracterizada por fiebre mayor de 38°C, artralgias en articulaciones interfalángicas proximales y muñecas, sin rigidez matutina y baja de peso no cuantificada. Tanto la fiebre como las artralgias eran de curso intermitente y no guardaban relación entre sí; presentaba, además, caída de cabello y prurito generalizado.
Debido a la persistencia de la sintomatología descrita y tener -hace un 1 año y medio- una prueba de anticuerpos antinucleares positiva con un título de 1:100, patrón moteado fino, asociado a anticuerpo anti-DFS70, fue diagnosticada de lupus eritematoso sistémico. Recibió tratamiento con prednisona 20 mg/día e hidroxicloroquina 400 mg/día, en esa ocasión presentó una prueba positiva de Coombs directo. Con el tratamiento indicado y luego de un año mostró remisión de los síntomas y se le disminuyó las dosis de los fármacos. Sin embargo, un mes antes de su ingreso reaparecieron los episodios de fiebre y artralgias, y agregó marcada baja de peso por lo que fue hospitalizada.
Al examen en hospitalización se le encontró adelgazada y pálida, además se palparon múltiples adenomegalias en región inguinal derecha, la mayor de 1cm, éstas fueron de consistencia dura, fijas y no dolorosas. En abdomen se encontró hepatomegalia de 14cm. En el examen locomotor se evidenció sinovitis en muñeca y en articulaciones interfalángicas proximales de mano derecha.
En el laboratorio, el hemograma mostró hemoglobina de 8,4 mg/dl, 13 660 leucocitos/mm3, 615 linfocitos/mm3 y plaquetas normales. Los valores de glucosa, urea y creatinina fueron normales, sin embargo, se encontró deshidrogenasa láctica en 2172 U/L, beta 2-microglobulina en 6,5 µg/ml y proteinograma electroforético con gammapatía policlonal. Las pruebas de ELISA para VIH y serologías para hepatitis B, HTLV I-II y sífilis fueron no reactivas. Los exámenes inmunológicos mostraron una prueba de Coombs directo (+), mientras que el factor reumatoideo, anticuerpos anti-péptido cíclico citrulinado y anticuerpos antinucleares fueron negativos, así como los valores de complemento C3 y C4 que también fueron normales. La baciloscopía de esputo fue negativa para bacilos ácido alcohol resistente en 3 oportunidades. El aspirado de médula ósea no fue significativo.
La tomografía de tórax y abdomen evidenció leve efusión pleural derecha y derrame pericárdico, además de múltiples adenomegalias mediastinales de hasta 12 mm, axilares de hasta 15 mm y periaórticas de hasta 5,6 mm, el hígado y bazo estaban aumentados de tamaño. La radiografía de manos comparativa no presentó signos de deformidad ni erosiones óseas.
Se procedió a la biopsia de la adenomegalia inguinal derecha de mayor tamaño. La histopatología mostró borramiento difuso de la arquitectura de los nódulos linfáticos y patrón folicular, con células atípicas alrededor de estructuras vasculares (Figura 1). La inmunohistoquímica además presentó CD3(+), CD4(+), PD-1(+), BCL6(+), CD20(-), CD23(+), EBV(+) y Ki67 40% (Figura 2). Con estos hallazgos y la insuficiente evidencia clínica y laboratorial de enfermedades difusas del tejido conectivo se hizo el diagnóstico de LAIT.
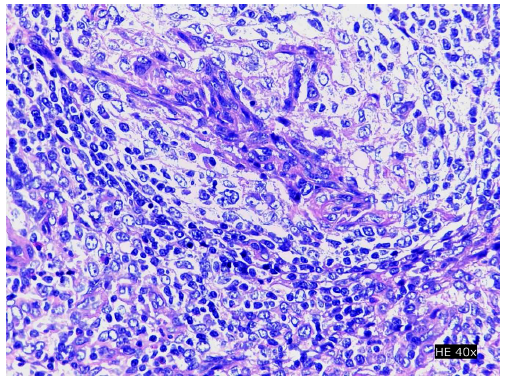
Figura 1 Estudio histopatológico de ganglio inguinal. Tinción de hematoxilina-eosina, 40X. Se aprecia presencia de linfocitos atípicos alrededor de estructuras vasculares.
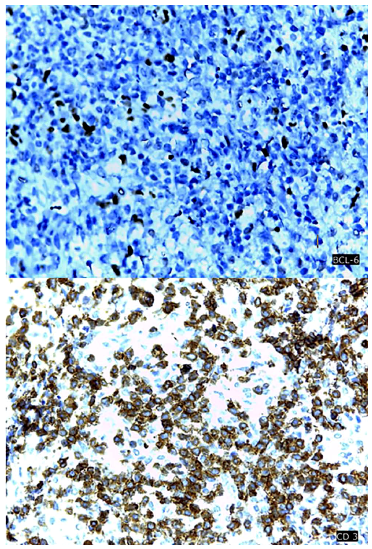
Figura 2 Inmunohistoquímica. Imagen superior con marcadores BCL-6 e imagen inferior con CD 3 positivos en muestra de ganglio inguinal.
La paciente recibió 3 sesiones de quimioterapia con dexametasona, ciclofosfamida, doxorrubicina, vincristina y etopósido, sin embargo, presentó cuadro de falla respiratoria aguda por neumonía y posterior falla multiorgánica con desenlace fatal.
Para el reporte de este caso, se obtuvo el consentimiento informado de parte de un familiar directo de la paciente, quien estuvo de acuerdo con lo reportado en el presente caso clínico. Además, se tuvieron los permisos institucionales para la revisión de la historia clínica.
DISCUSIÓN
El LAIT es un subtipo de los linfomas de células T periféricos derivado de células T colaboradoras foliculares. El LAIT representa el 15 al 20 % de los linfomas periféricos de células T y el 1 al 2 % de los linfomas no Hodgkin, afecta principalmente a la población de edad avanzada, siendo más frecuente entre 60 y 65 años, con una ligera mayor frecuencia en hombres 2,4.
Sus manifestaciones clínicas son variables y no específicas, los síntomas B aparecen en más del 70 % de los casos. La presencia de fiebre, sudores nocturnos, adenomegalias periféricas y generalizadas, hepatoesplenomegalia, baja de peso, erupciones cutáneas, artralgias y artritis, además de efusión pleural y ascitis son las manifestaciones más frecuentes 2,4. Dicho cuadro clínico puede confundirse -al inicio- con enfermedades difusas del tejido conectivo como lupus eritematoso sistémico 5, artritis reumatoide 6, esclerosis sistémica 7, enfermedad de Still del adulto 8 y síndrome de Sjögren 9.
La afectación de los linfocito T CD4+ en este tipo de linfoma, que en condiciones normales regulan la producción de anticuerpos llevaría a una desregulación inmune, lo que originaría las diversas manifestaciones autoinmunes 4,10. Nuestra paciente tenía una edad inusual para la presentación de este tipo de linfoma y había sido diagnosticada 1 año y medio antes de lupus eritematoso sistémico. Este diagnóstico fue hecho en base a la presencia de alopecia, fiebre, artralgias y baja de peso, sumado a la positividad de anticuerpos antinucleares y test de Coombs directo positivo, hallazgos de laboratorio que pueden observarse en el LAIT (Tabla 1) 3,11,12.

Precisamos que los anticuerpos antinucleares de la paciente estuvieron asociados a la positividad del anticuerpo an-ti-DFS 70, el cual es descrito en personas sanas y su evidencia aislada no debería inducir al diagnóstico de enfermedades difusas del tejido conectivo, más aún si no existe otro marcador de autoinmunidad ni cuadro clínico sugerente 13. En nuestro caso y debido a las manifestaciones clínicas descritas, se hizo el diagnóstico inicial de lupus eritematoso sistémico.
Dentro del laboratorio actual se repitió la prueba de anticuerpos antinucleares la cual fue negativa, sin embargo, se encontró elevación de deshidrogenasa láctica y beta 2 microglobulina. Además se encontró linfopenia, anemia moderada, prueba de Coombs directo positiva y gammapatía policlonal, lo que puede encontrarse tanto en casos de enfermedades linfoproliferativas como autoinmunes 2,5,12. Otras alteraciones que pueden encontrarse en este tipo de linfoma y que no se encontraron en nuestro caso incluyen la presencia de crioglobulinas, crioaglutininas, eosinofilia, trombocitopenia, disfunción tiroidea y en 70% compromiso de médula ósea 2,14.
Al haber duda diagnóstica y tal como se recomienda en estos casos, se procedió a una biopsia de ganglio inguinal. La histopatología de nuestro caso fue compatible con LAIT, al describirse borramiento de la arquitectura del ganglio linfático, con células atípicas por fuera de los folículos y alrededor de estructuras vasculares. Además en la inmunohistoquímica se encontró expresión de CD3(+), CD4(+), PD1(+), BCL-6(+), CD23(+) y Ki67 40 %, lo cual es característico de la enfermedad 2,14.
En la inmunohistoquímica se encontró además positividad para virus de Epstein-Barr, el cual se ha descrito en un 80-90 % de casos de LAIT 4, si bien su presencia no tendría relación con la formación del linfoma 2,15, se ha reportado que una viremia alta por este virus al momento del diagnóstico se ha asociado a un peor pronóstico 14. Otros hallazgos fueron el marcador BCL-6(+), el cual se expresa en linfomas que tienen como estirpe al linfocito T colaborador y el marcador PD-1(+), el cual se describe hasta en un 95 % en casos de LAIT 3.
Realizado el diagnóstico se inició el tratamiento con el esquema ciclofosfamida, doxorrubicina, vincristina y dexametasona (esquema CHOP), asociado a etopósido, el cual ha demostrado en algunas series una tasa de respuesta inicial de hasta 82 %, sin embargo, la remisión se reporta como no duradera, no teniéndose al momento una terapia establecida para el LAIT debido a su poca frecuencia 14,16. En nuestra paciente el desenlace fue fatal debido a un cuadro de insuficiencia respiratoria aguda secundaria a neumonía.
El curso clínico del LAIT generalmente es desfavorable. El LAIT es diagnosticado en estadios avanzados en un 80 % de los casos 14, y tiene un promedio general de sobrevida a los 5 años de 30 % 4, sin embargo, también hay reportes de mayor esperanza de vida desde que se comenzó a estudiar esta enfermedad 11.
En conclusión, el LAIT es un reto diagnóstico debido a que puede imitar varias patologías autoinmunes como el lupus eritematoso sistémico, es por ello, que recomendamos que ante la evidencia de adenomegalias en pacientes con enfermedades difusas del tejido conectivo se proceda a la biopsia para, de ser el caso, realizar un diagnóstico y tratamiento precoces que mejoren la sobrevida de los pacientes.

