











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La leishmaniasis cutánea (LC) es la forma más común de la enfermedad causada por Leishmania spp. El riesgo del incremento de LC es latente debido a que el cambio climático afecta la distribución de los flebótomos y la migración de las poblaciones a regiones con alta transmisibilidad 1. Leishmania braziliensis es el principal agente etiológico de LC y mucosa en América del Sur, el promastigote meta-cíclico ingresa a las células diana mediante receptores, siendo fundamentales los de tipo Toll (TLR).
La señalización dependiente de TLR ocasiona el incremento de la respuesta celular innata mediada por la producción de citoquinas proinflamatorias. Durante la etapa inicial de la infección, las células hospedadoras liberan especies reactivas de oxígeno y nitrógeno, cuya intensidad no está relacionada con la carga parasitaria 2. Aunque la mayoría de los estudios consideran la función protectora de los TLR, el hecho de que los parásitos se aprovechen de estos receptores promueve la infección 3. El objetivo del estudio fue cuantificar y comparar la producción de óxido nítrico y los niveles de transcripción de TLR-3, TLR-9 (endosomales), TLR-4, IL-12 y TNF-α entre macrófagos murinos infectados y no infectados con la especie nativa de Leishmania sp. (Lbn).
MÉTODOS
Identificación de Lbn
La cepa fue donada en estadio de promastigote (pasaje 234, aislada de exudado de LC) por el Laboratorio de Zoonosis del Instituto Nacional de Salud (INS), Lima, Perú. Su mantenimiento se realizó en medio bifásico elaborado con sangre desfibrinada humana al 10% (37 °C, 5% CO2, 95% humedad relativa). Para la identificación de la especie se extrajo el DNA a partir de ~3,2 x 105 promastigotes/mL empleando QIAamp DNA Mini Kit (Qiagen) según las indicaciones del fabricante. Los primers (Inmunochem) diseñados para Leishmania (Viannia) braziliensis4 fueron analizados empleando BLAST seguido de Oligoanalyzer Tool. Las secuencias fueron: F (Forward) 5´ AAATTCGCGTTTTTTGGCCTCCCCG 3’, posición en el genoma: 185-209, Tm: 66.2 °C; R (Reverse) 5´GCATAAACTAGAGACGGAACAGAG 3´, posición en el genoma: 643-620, Tm: 62.9 °C.
La amplificación se hizo por qPCR empleando minicírculos de kDNA. Se elaboró una mezcla formada por 10 ng de DNA total, 0,5 μM de cada primer, 2,5 mM de los dNTPs (Invitrogen), 2 mM MgCl2, Taq polimerasa 2,5 µL/ reacción y buffer de PCR 10X (sin MgCl2) (Invitrogen) en un volumen final de 25 µL. Los pasos del qPCR fueron: desnaturalización inicial (94 °C, 15 min), desnaturalización final (94 °C, 1 min), alineamiento (55 °C, 30 s, 40 ciclos), extensión inicial (72 °C, 1 min) y extensión final (72 °C, 7 min). Los amplicones se visualizaron mediante electroforesis horizontal en agarosa (1,5%, 120 V, 40 min) y fotografiaron en un transiluminador (Bio-Rad). Se empleó un marcador de 100-1000 pb (Sigma).
La purificación del amplicón se hizo con el kit QIAquick-Gel Extraction, seguido del kit Big Dye Terminator Cycle Sequencing Ready Reaction (Perkin-Elmer), utilizando los primers respectivos (F y R) siguiendo las indicaciones del fabricante. El secuenciamiento se hizo en las siguientes condiciones: activación inicial a 94 °C (3 min), 24 ciclos de 30 s (96 °C), 30 s (51°C) y 4 min (60 °C). Para el secuenciamiento se utilizó ABI PRISM 310 Genetic Analizer (Applied Biosystems). Para encontrar la secuencia consenso se utilizó el programa CLUSTAL X versión 1.83. Posteriormente, estas secuencias se contrastaron con otras del GenBank utilizando BLAST (www.blast.ncbi.nlm.nih. gov) y se analizaron con el software Seq-Man (Dnastar Inc., Wisconsin, USA). Para el análisis filogenético se empleó MEGA versión 5.10 y el método Neighbor Joining, Tamura-Nei de 1000 repeticiones.
Obtención de macrófagos peritoneales
Se inocularon 1,5 mL de caldo tioglicolato de Brewer por vía intraperitoneal (ip) a ratones Balb/c adquiridos del INS (n=6, hembras de 8 semanas de edad). Después de 3 días, se inoculó 1 mL de RPMI (ip, 15 min). Los ratones fueron sedados en cámara de cloroformo y eutanizados por dislocación cervical. Los macrófagos se lavaron 2 veces con PBS 1X (10 °C, 1000 rpm, 7 min), el pellet se resuspendió en 3 mL de RPMI 1640 10% de suero de bovino fetal, antibióticos y antimicótico (coRPMI, Gibco), se colocaron en placas Petri e incubaron (1 h, 33 °C, 5% CO2), se lavó con PBS 1X, se añadieron 2 mL de coRPMI, los macrófagos adheridos se colectaron y se centrifugó a 2000 rpm (3 min). El pellet se resuspendió en coR-PMI. Dependiendo del tipo de ensayo, se cultivaron en microplacas de 96 pocillos con fondo plano o laminillas cubreobjetos (37 °C, 5% CO2).
Infección de macrófagos con Lbn
La fase líquida del medio bifásico (promastigotes estacionarios) se colectó y centrifugó (1000 rpm, 5 min), el pellet se re-suspendió en coRPMI. Para la infección se ajustó la concentración en la proporción de 1 macrófago: 10 promastigotes 5. Los co-cultivos (macrófago-Lbn) se hicieron sobre laminillas, en cámara húmeda y se incubaron durante 24 y 48 h, los controles fueron macrófagos no infectados (n = 3). Las laminillas se colorearon con Giemsa y analizaron por microscopía óptica. El porcentaje de macrófagos infectados (MI) en los co-cultivos se determinó contando 100 macrófagos con o sin amastigotes intracelulares 6 (n = 3). La capacidad infectiva fue representada como el resultado del valor promedio de amastigotes en MI multiplicado por el porcentaje de MI. El valor promedio de amastigotes se determinó mediante el conteo de éstos por cada 100 MI 7.
Producción de óxido nítrico
Después de centrifugar, se colectaron los sobrenadantes de los co-cultivos y controles a las 0, 2, 12, 15, 24 y 48 h de incubación (n = 3). A 100 µL del sobrenadante se agregaron 100 µL del reactivo de Peter Griess (1 min) y se midió la absorbancia (540 nm). Para la expresión transcripcional (ET) de TLRs y citoquinas, se emplearon co-cultivos de 24 h de incubación 8 (n = 3).
Obtención de RNA para la expresión transcripcional de TLRs y citoquinas
La extracción de RNA se hizo con el kit Power SyBr Green Cell-To TM (Applied Byosistem), siguiendo las indicaciones del fabricante. La concentración determinada se utilizó como referencia de la cantidad aproximada de RNA para qRT-PCR.
Retrotranscripción a DNA complementario (cDNA) de TLRs y citoquinas
Se empleó el kit Power SyBr Green Cell-T o TM siguiendo las indicaciones del fabricante. Para la síntesis de cDNA se incubó a 37 ºC (60 min), y para la desnaturalización de la enzima a 95 ºC (5 min). El cDNA se almacenó a -20 °C. En la tabla 1 se presentan las secuencias de los primers9. Como control endógeno se utilizó GAPDH.

Amplificación de cDNA para TLRs y citoquinas
La cuantificación de la expresión de los primers TLR-3, TLR-4 y TLR-9 se hizo con el kit Master Mix Power SyBrGreen PCR (Applied Byosistem) y la amplificación de cDNA (qRT-PCR) con el Mix de SyBr Green (Thermocycler Corbett). Se utilizaron 5 µL de cDNA diluido en agua ultra pura (1:3 respectivamente). La concentración final de los primers fue de 500 nM y de cDNA de 2,7 µg/µL. Como control negativo se utilizó agua ultrapura (n = 3). Las condiciones de PCR para los tres primers fueron: 95 °C (10 min) para activar la enzima, 95 °C (15 s) y 53 °C (1 min) para los 40 ciclos de PCR y calentamiento gradual cada 0,1 °C, desde 50 hasta 99 °C para la curva de disociación. La temperatura media para la amplificación fue de 52 °C (TLR-3), 51, 6 °C (TLR-4), 50, 4 °C (TLR-9) y 54, 9 °C (GAPDH). En el caso de las citoquinas, la temperatura media para la amplificación fue de 50,5 °C (IL-12), 55,4 °C (TNF-α) y 54,9 °C (GADPH). La cuantificación relativa para TLRs y citoquinas fue calculada mediante Delta-Delta Ct con el programa de Applied Biosystems y la fórmula 2-∆∆CT.
Análisis estadístico
La media aritmética y la desviación estándar de la producción de ON se determinó con Microsoft Excel 2016. Para la significancia se utilizó la prueba t de Student de muestras independientes y se consideró significativo si p < 0,05. La ET se calculó con el programa SPSS (versión 15).
RESULTADOS
En el análisis de las secuencias obtenidas para Lbn, el amplicón tuvo 536 pb (Figura 1A- primer F). Se obtuvo un producto de 393 nucleótidos. Al confrontar las secuencias de Lbn con L. (V.) braziliensis se encontró una homología de 98% con un valor de E de 0,0 (M87315.1).
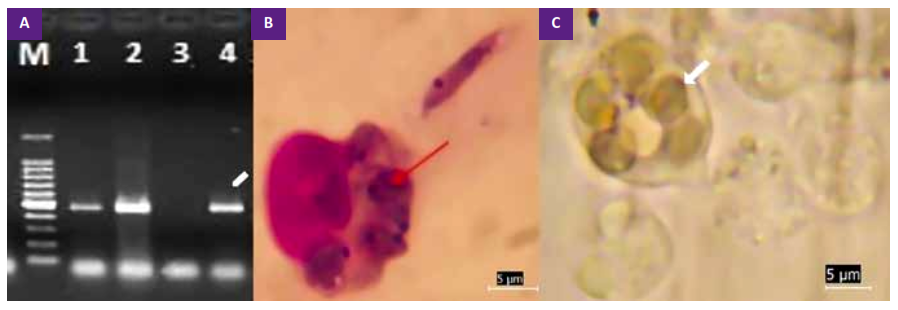
Figura 1. (A) Amplicón de kDNA de promastigotes de L. braziliensis Lbn, la flecha señala el amplicón para F (536 pb, carriles 1, 2, 4). M: Marcador molecular de PCR (100-1000 pb), control negativo (carril 3). (B) Macrófago infectado con L. braziliensis Lbn (24 h), la flecha señala amastigotes, externamente al macrófago se observa el promastigote (Giemsa, x 1000). (C) Macrófago con vacuolas parasitóforas después de 6 h de co-cultivo (fresco, x 1000).
Se colectaron 1,83 x 105/mL macrófagos murinos infectados con Lbn por ratón. Después de 24 h de co-cultivo, el porcentaje de MI fue (~13,38%, a las 48 h se incrementó a (~37,47%. En cuanto a los amastigotes, a las 24 h de co-cultivo fue de (~3,1 y a las 48 h fue (~5,3 (Figura 1B). La fagocitosis estuvo acompañada por la mayor formación de vacuolas parasitóforas (Figura 1C). La capacidad infectiva se incrementó de 40,14% (24 h) a más del 100% (48 h) (p < 0,001).
La producción de óxido nítrico en los MI fue mayor a las 2 h (~2,53 μM), mientras que en los controles fue de (~1,54 μM (p < 0,05). En los controles se incrementó en función al tiempo, llegando a (~13,69 μM a las 48 h (p < 0,0001) (Figura 2).
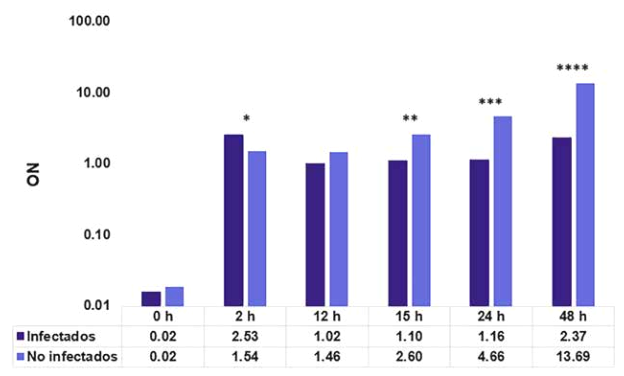
Figura 2. Cuantificación de óxido nítrico (ON). En los cultivos de macrófagos infectados con L. V. braziliensis la mayor producción de ON se presentó a las 2 h. Las diferencias se determinaron mediante t - Student para muestras independientes, (n = 3). *p < 0,05, **p < 0,01, ***p < 0,001, ****p< 0,0001.
La expresión transcripcional (ET) para TLR-3 fue 5,12 veces mayor (p < 0,001), TLR9 fue 3,95 veces (p < 0,01) y TLR-4 fue 3,23 veces (p < 0,05), respecto a los controles (Figura 3A). La diferencia de niveles de ET entre TLR-4 y TLR-9 no fue significativa (p = 0,12). También, se encontró 4,18 veces mayor nivel transcripcional para las dos cito-quinas proinflamatorias en los MI respecto a los controles (p < 0,001). En el caso de TNF-ɑ fue 3,66 veces mayor (p< 0,001) (Figura 3B).
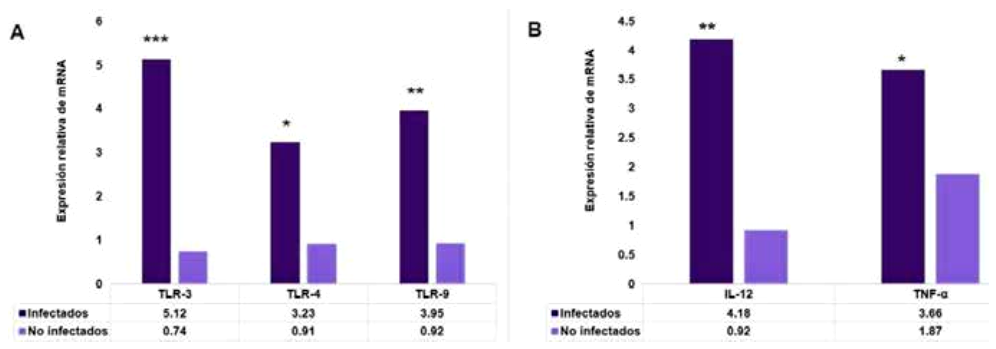
Figura 3. Expresión transcripcional de TLR y citoquinas proinflamatorias en macrófagos infectados con L. braziliensis Lbn. (A) Se encontró mayor nivel transcripcional para los TLR-3, TLR-4 y TLR-9 respecto a los no infectados (controles). (B) Se determinó mayor nivel transcripcional para IL-12 y TNF-ɑ respecto a los controles. Se aplicó t-Student para muestras independientes (n = 3). *p < 0,05, **p < 0,01, ***p < 0,001.
DISCUSIÓN
La cepa Lbn mostró una homología del 98% para L. (V.) braziliensis coincidiendo con Satow et al.11. El amplicón tuvo 536 pb (cinetoplasto) para la misma especie se reportó un amplicón de 599 pb (citocromo B) 12. Los macrófagos peritoneales primarios se consideran maduros y más estables en su funcionalidad y fenotipo 8,13 y permitieron verificar el incremento de la capacidad infectiva de Lbn. El número de amastigotes por macrófago a las 48 h (~5,3 amastigotes) fue mayor que el reportado 5 días posinfección (Figura 1A, 1B y 1C) 14, diversos factores influyen sobre estos valores 7.
La producción de ON por los MI fue significativa únicamente a las 2 h posinfección (Figura 2), el incremento en los controles podría estar relacionado con algún componente del coRPMI 1640 15; asimismo, la baja producción sugiere que Lbn es resistente al ON 16 o existe mayor consumo de glucosa 17.
En los MI, la mayor expresión transcripcional encontrada para TLR-3, seguida de TLR-9 y TL-4, y los niveles elevados de IL-12 y TNF-ɑ (Figuras 3A y 3B) confirmarían que la participación de TLR-9, TLR-4, TLR-2 y TLR-3 es fundamental para generar una respuesta de citoquinas proinflamatorias 19, y especies reactivas de oxígeno y nitrógeno 19,20. La menor expresión transcripcional para TLR-4 podría estar relacionada con la baja producción de ON 18.
La expresión transcripcional de TLR-3 resultó predominante indicando mayor estimulación intracelular. Se han observado fuertes interacciones de TLR-2 y TLR-4 con las formas clínicas asociadas a L. (V.) braziliensis, mientras que TLR-9 lo hizo con L. (L.) amazonensis21, para esta especie se demostró que el TLR-3 es fundamental en el crecimiento intracelular y supervivencia del parásito 22. No se encontró información acerca de TLRs en MI, animales o humano, infectados con L. braziliensis nativas en Perú.
La expresión transcripcional para IL12 y TNF-ɑ indican que la infección por L. V. braziliensis promueve una potente respuesta celular 23, corroborada por la reducción del estrés oxidativo, número de células infectadas y citoquinas proinflamatorias cuando se bloquean TLR-2 y TLR-4 24. Esto sugiere que los antagonistas y agonistas de TLR pueden ser excelentes inmunomoduladores 21.
El estudio tiene limitaciones como no haber incluido una cepa referencial, un inductor de citoquinas proinflamatorias y un ligando específico para el TLR a evaluar, a fin de determinar si existen diferencias en la ET de estos receptores y las citoquinas proinflamatorias entre cepas nativas y referenciales.
En conclusión, se verificó la activación del macrófago por señales de peligro (Lbn) tanto a nivel extracelular (TLR-4) como intracelular (TLR-3 y TLR-9) directamente relacionada con la significativa ET para IL-12 y TNF-ɑ. El modelo podría ser utilizado para evaluar fármacos y productos agonistas y antagonistas en LC.

