










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista de Investigaciones Veterinarias del Perú
Print version ISSN 1609-9117
Rev. investig. vet. Perú vol.29 no.2 Lima Apr./Jun. 2018
http://dx.doi.org/10.15381/rivep.v29i2.14523
ARTÍCULOS PRIMARIOS
Desarrollo de una plataforma molecular para la detección y cuantificación del virus vacunal de Newcastle
Development of a molecular platform for the detection and quantification of Newcastle vaccine virus
Phillip Ormeño-Vásquez1, Luis Tataje-Lavanda1, Katherine Huamán-Gutiérrez1, Katherine Calderón1, Vladimir Longa-Bobadilla1, Ángela Montalván1, Edison Huaccachi-Gonzáles1, Manuel Criollo-Orozco1, Jorge Bendezú1, Manolo Fernández-Díaz1
1 Laboratorios de Investigación y Desarrollo de Farmacológicos Veterinarios (FARVET S.A.C.), Carretera Panamericana Sur N° 766 Km 198.5, Chincha Alta, 11702, Ica,Perú
2 E-mail: phillip.ormeno@farvet.com
RESUMEN
El estudio tuvo por objetivo desarrollar una plataforma molecular para la cuantificación del virus de la enfermedad de Newcastle (NDV) a partir de un sistema de cultivo en huevos embrionados SPF. Primero se evaluaron cuatro pares de cebadores que amplifican diferentes regiones del genoma viral de NDV que codifican para la proteína de nucleocápside (NP), proteína matriz (M), proteína fusión (F) y la ARN polimerasa ARN dependiente (L), con la finalidad de seleccionar el más conservado a partir del cual se desarrolló una plataforma molecular basada en la transcripción reversa -reacción en cadena de polimerasa convencional (RT-PCRc) en dos pasos para la detección del NDV. Posteriormente, esta fue llevada a transcripción reversa -reacción en cadena de polimerasa en tiempo real (RT-qPCR) para la cuantificación de NDV producido a partir de un sistema de huevos embrionados. Mediante estas técnicas se determinó que los cebadores para el gen M fueron adecuados según los criterios de optimización para el desarrollo de ambos métodos. Mediante ensayos de sensibilidad se demostró que la RT-qPCR (116 copias genómicas/µl) era 10 veces más sensible que el RT-PCRc. Los cebadores fueron específicos pues no hubo amplificados en los controles negativos ni en otros patógenos aviares (virus de la laringotraqueitis infecciosa, metapneumovirus aviar, virus de la bronquitis infecciosa, Avibacterium paragallinarum, Gallibacterium anatis y Ornithobacterium rhinotracheale). Debido su sensibilidad y especificidad, se propone esta plataforma para la cuantificación de NDV vacunal cuando es producido a partir de un sistema de huevos embrionados, como una alternativa frente a métodos convencionales de titulación como hemaglutinación, ensayo en placa, TCDI50 y DIEP50.
Palabras clave: NDV; RT-PCRc; RT-qPCR; huevos embrionados SPF
ABSTRACT
The objective of the study was to develop a molecular platform for the quantification of Newcastle disease virus (NDV) from a culture system in embryonated SPF eggs. First, four pairs of primers were evaluated that amplify different regions of the NDV viral genome that code for: nucleocapsid protein (NP), protein matrix (M), fusion protein (F) and RNA-dependent RNA polymerase (L) to select the most conserved one from which a molecular platform based on reverse transcription was developed -conventional polymerase chain reaction (RT-PCRc) in two steps for the detection of NDV. Subsequently, it was taken to reverse transcription -real-time polymerase chain reaction (RT-qPCR) for the quantification of NDV produced from a system of embryonated eggs. Through these techniques, it was determined that the primers for the M gene were adequate according to the optimization criteria for the development of both methods. Sensitivity tests showed that the RT-qPCR (116 genomic copies/µl) was 10 times more sensitive than the RT-PCRc. The primers proved to be specific since there were no amplifications in the negative controls or in other avian pathogens (infectious laryngotracheitis virus, avian metapneumovirus, infectious bronchitis virus, Avibacterium paragallinarum, Gallibacterium anatis and Ornithobacterium rhinotracheale). Due to its sensitivity and specificity, this platform is proposed for the quantification of NDV vaccine when it is produced from an embryonated egg system, as an alternative to conventional titration methods such as hemagglutination, plaque assay, TCDI50 and DIEP50.
Key words: NDV; RT-PCRc; RT-qPCR; embryonated SPF eggs
INTRODUCCIÓN
El virus de Newcastle (NDV, de sus siglas en inglés Newcastle disease virus) es el agente causal de la enfermedad de Newcastle (ND), enfermedad altamente contagiosa con un amplio rango de huéspedes y reportada en todos los órdenes de aves (Seal et al., 1995). ND está distribuida a nivel mundial, donde es causante de notables pérdidas económicas para el sector avícola, y en el Perú está presente como enfermedad de reporte obligatorio (OIE, 2017). El NDV es un miembro de la familia Paramixoviridae, un virus envuelto que tiene un genoma de una sola hebra de ARN lineal de polaridad negativa. El genoma de ARN tiene un tamaño aproximado de 15 200 pb y contiene seis genes estructurales que en orden 3´ a 5´ del ARN genómico codifican, la proteína de nucleocápside (NP), fosfoproteína (P), proteína matriz (M), proteína fusión (F), hemaglutinina-neuramidasa (HN) y ARN polimerasa ARN dependiente (L) (Cattoli et al., 2011; Alexander et al., 2012).
NDV puede ser clasificado en cepas altamente patógenicas (velógenico), intermedias (mesogénica) y no patogénicas (lentogénicos) basados en su patogenia en pollos (Cattoli et al., 2011). La mejor estrategia de prevención es la vacunación con cepas atenuadas, la cual ha sido ampliamente usada por más de 50 años (Gallili y Ben-Nathan, 1998). Actualmente, la producción de vacunas de NDV es principalmente realizada en sistemas de huevos embrionados libres de patógenos específicos (SPF) debido a su accesibilidad y bajo costo (Seene et al., 2003); por otra parte, la medición del título exacto es importante en la producción de vacunas virales, valiéndose de ensayos de hemaglutinación, ensayos en placa y de dosis media infectiva en tejidos (TCDI50) y embriones (DIEP50). Sin embargo, estos métodos pueden no ser altamente específicos y consumir un tiempo considerable (McGinnes et al., 2006).
La técnica de qPCR tiene muchas ventajas sobre los métodos tradicionales, incluyendo rapidez, posibilidad de cuantificación, alta sensibilidad y especificidad (Jang et al., 2011). Varios métodos de RT-qPCR han sido desarrollados para detectar y cuantificar NDV (Seal et al., 1995; Tan et al., 2009; Fuller et al., 2010; Jang et al., 2011). Sin embargo, la mayoría de estos métodos están enfocados en el diagnóstico de aves infectadas con ND, pero no todos incluyen criterios de optimización específicos. Por tanto, se requiere optimizar una RT-PCR que pueda ser utilizada para cuantificar el NDV utilizado en la producción de vacunas aviares.
Este estudio tuvo por objetivo desarrollar una plataforma molecular para la cuantificación del NDV a partir de un sistema de cultivo en huevos embrionados SPF. Para lograr esto, se amplificaron, a través de RT-PCRc, diferentes regiones del genoma viral utilizando cebadores dirigidos hacia los genes F, NP, L y M del NDV. Los cebadores que cumplieron con las características/criterio de selección (Innis y Gelfand, 1999; Ottiger, 2010) fueron utilizados para cuantificar el virus a través del RT-qPCR.
MATERIALES Y MÉTODOS
Cepas Virales
La cepa vacunal NDV LaSota forma parte del cepario de los Laboratorios FARVET (Perú), la cual fue reactivada en huevos embrionados de pollo SPF de 9 días de edad por ruta alantoidea. Luego el fluido alantoideo fue centrifugado a 3000 g por 15 min y el sobrenadante fue almacenado en alícuotas a -80 °C hasta su uso. El ARN genómico del virus de la bronquitis infecciosa aviar (IBV), Metapneumovirus aviar (SHS) y el ADN genómico del virus de la virus de la laringotraqueitis infecciosa (ILTV), y de las bacterias Avibacterium paragallinarum, Gallibacterium anatis y Ornithobacterium rhinotracheale forman parte del cepario de los Laboratorios FARVET SAC.
Cebadores
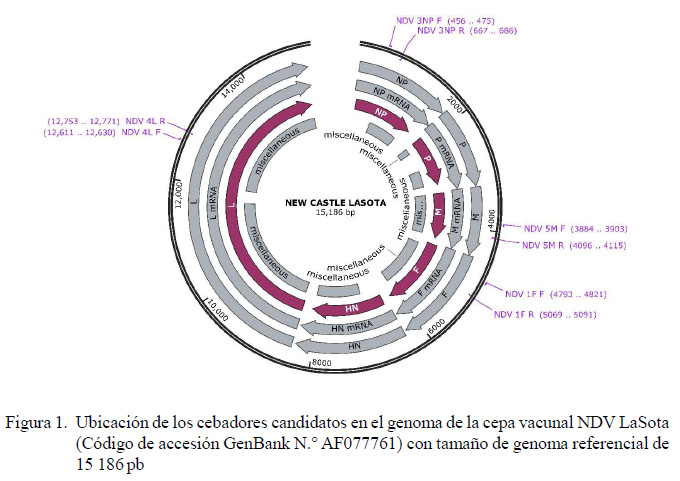
Se seleccionaron cebadores candidatos dirigidos a las regiones F, NP, L y M del virus NDV LaSota, en base a revisión bibliográfica (Seal et al., 1995; Farkas et al., 2009; Fuller et al., 2010; Cattoli et al., 2011). El criterio de inclusión fue la capacidad de detectar, por lo menos cepas de NDV de Clase I. Los cebadores fueron sintetizados de forma comercial (IDT, EEUU). Las secuencias de los cebadores y otros detalles están descritas en el Cuadro 1. Los cebadores fueron denominados siguiendo la abreviatura del virus, el gen al cual fue dirigido y un número. Se comprobó su ubicación en la cepa vacunal utilizada, empleando el programa SnapGene Viewer v. 3.3.3 (GSL Biotech, EEUU) (Figura 1).

Producción y Purificación de NDV
Se infectaron huevos embrionados SPF de 9 días con la cepa NDV LaSota a una multiplicidad de infección (MOI) de 0.01 e incubados a 37 °C durante 96 horas. El sobrenadante fue cosechado y centrifugado a 3000 g durante 30 min a 4 °C para eliminar el debris celular. Luego, fue pasado por un filtro de 0.22 µm. El título de NDV del fluido alantoideo fue determinado por hemaglutinación (McGinnes et al., 2006; OIE, 2017) y usado como inóculo para posteriores experimentos.
Para la purificación del virus, el fluido alantoideo de NDV previamente clarificado fue ultracentrifugado en botellas de 250 ml en el rotor Type 19 de la ultracentrífuga Optima L-100K (Beckman Coulter, EEUU) a 48 000 g durante 18 horas. El sobrenadante fue descartado y los pellets fueron resuspendidos en 5 ml de PBS 1X. Luego, fueron transferidos a tubos de policarbonato previamente llenados con sucrosa al 30%(P/V) (Sigma Aldrich, EEUU), centrifugados en el rotor SW28 a 11 000 g durante 18 horas. Se descartó el sobrenadante y el pellet viral fue usado para el aislamiento de ARN de NDV.
Aislamiento de ARN Genómico Viral
El pellet viral fue extraído con el RNeasy Midi Kit (Qiagen, Alemania), de acuerdo con las instrucciones del fabricante. Los productos obtenidos se conservaron a -80 °C hasta su uso.
Preparación de Material Genómico de NDV LaSota
El ARN genómico de cadena sencilla (ssARN) de NDV LaSota fue sometido a diluciones seriadas desde 1 ng hasta 100 ag por cada µl de ARN genómico, procedimiento que también se siguió para el cADN obtenido por retrotranscripción reversa que fue llevada a cabo con el kit comercial ProtoScript® First Strand cDNA Synthesis Kit (New Englands Biolabs, UK) en el termociclador MasterCycler Pro-S (Eppendorf, Alemania). El cADN obtenido se cuantificó por fluorometría en el Qubit (Invitrogen, EEUU). La pureza fue determinada por la relación A260/A280 en el Biophotometer plus (Eppendorf, Alemania) para ambos tipos de ácidos nucleicos.
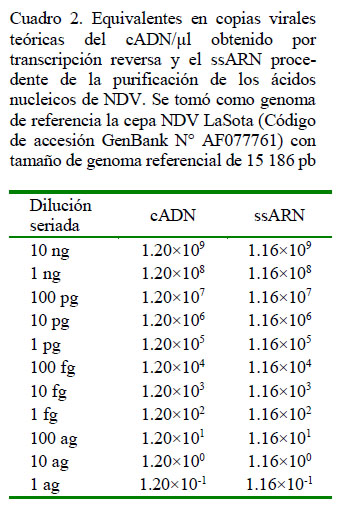
La cuantificación de NDV presente en el fluido alantoideo fue llevada a cabo en el Qubit 1.0 Fluorometer (Invitrogen, EEUU) y para determinar los equivalentes en copias virales teóricas se usó la siguiente fórmula (Ke et al., 2006): Y = (moléculas/µl)=X (g/l) (tamaño del genoma x 340) x 6.02 x 1023 para el material de ARN y la formula Y = (molé-culas/µl)=X (g/l) (tamaño del genoma x 330) x 6.02 x 1023 para material de cDNA. Con base a estos valores se construyó una tabla de equivalencias (Cuadro 2).
Optimización de la RT-PCR Convencional
El RT-PCRc fue desarrollado en dos pasos: la transcripción reversa para la obtención cADN fue llevada a cabo con el Kit ProtoScript (New England Biolabs, EEUU) de acuerdo con las instrucciones del fabricante. Brevemente, en un volumen final de 20 µl usando 5 µl de ARN total, 2 µl de Ramdom hexamer primer (600 µM), 2 µl de ditiotreitol (100 µM), inhibidor de ARNasa murina (40 u/µl) y 200 U de MMLV (transcriptasa reversa del virus de la leucemia Murina de Moloney). La retrotranscripción se llevó acabo en un termociclador Mastercycler®Pro-S (Eppendorf, EEUU) a 25 °C durante 5 min y posteriormente a 42 °C durante 3 horas. Luego se cuantificó por fluorometría (Qubit 1.0 Invitrogen, EEUU) y se ajustó la concentración de cDNA deseada tomando al final 2 µl para la PCR que se realizó en el termociclador mencionado. Se empleó el kit GoTaq Flexi DNA-Polimerasa (Promega, EEUU), con deoxyribonucleósido trifosfato (dNTPs) a una concentración de 200 µM, MgCl2 2.0 mM, GoTaq DNA Polimerasa (1.25 u) y GoTaq Flexi Buffer 1x. La denaturación inicial a 94 °C x 5 min, 35 ciclos de 90 °C x 30 s, (45-65 °C) x 30 s, 72 °C x 30 s y un paso de extensión final a 72 °C por 5 min. Se optimizaron las condiciones de PCR, donde se evaluó temperatura de hibridación y sensibilidad para la totalidad de los cebadores candidatos y, luego, al par de cebadores con las condiciones ideales se ajustó la concentración de MgCl2 y de los cebadores (Innis y Gelfand, 1999). El cADN de fluido alantoideo de huevos SPF fue utilizado como control negativo y el agua de grado PCR fue utilizada como blanco, los cuales fueron incluidos en cada corrida.
Los productos de PCR fueron corridos en un sistema de electroforesis horizontal Compact M (Analityk Jena, Alemania). Los geles fueron preparados al 1.5% con agarosa (Cleaver Scientific, UK), buffer TAE y SyberSafe (Invitrogen, EEUU) electroforetizados a 80 mV durante 75-90 min y visualizados en un transiluminador de luz azul Safe Imager 2.0 (Invitrogen, EEUU). Se emplearon los marcadores de peso molecular 100 bp y O’Gene Ruler (Thermo Scientific, EEUU).
Optimización de la RT-qPCR en un Paso Usando Sybr Green I
En base al par de cebadores del gen candidato que cumplió con los parámetros optimizados de temperatura de hibridación, sensibilidad, concentración de cebadores, concentración de MgCl2 y especificidad, se desarrolló el RT-qPCR utilizando el kit KAPA SYBR FAST One-step for LightCycler 480 Mix (Kapa Biosystems, EEUU), donde cada reacción de PCR fue llevada a cabo tomando ARN genómico purificado. El protocolo de RT-qPCR consistió en 40 ciclos de 95 °C durante 10 s, hibridación de 60 °C por 20 s y extensión de 72 °C por 20 s. Las curvas de disociación consistieron en 95 °C por 5 s, 65 °C por 30 s y calentado hasta 97 °C a una velocidad de 0.1 °C/s. Los productos de amplificación fueron detectados mediante análisis de curva de disociación y confirmados por secuenciación (Macrogen, Corea). Los valores del ciclo de amplificación (Cp) y temperatura de disociación (Tm) fueron analizados con el Software LightCycler 480 v. 1.5.0. Los análisis estadísticos fueron realizados con el programa Microsoft Excel v. 2013.
Ensayos de Sensibilidad
Para los ensayos de sensibilidad se realizaron diluciones decimales seriadas de cADN/ssARN para la RT-PCRc y RT-qPCR, respectivamente, que fueron desde 10 ng hasta 1 ag/µl. Los ensayos se realizaron por triplicado durante tres días consecutivos. Para la RT-PCRc se empleó el termociclador MasterCycler (Eppendorf,Alemania), y para la RT-qPCR se usó el temociclador Light Cycler 480 (Roche, EEUU). Cada dilución fue corrida hasta 10 veces durante 3 días y se determinó la sensibilidad como la dilución previa a aquella que poseía un coeficiente de variación >5%. Se tomó en cuenta la presencia de banda de interés o la formación de curvas de fluorescencia con valor Ct <35 y/o la presencia de pico de disociación para el RT-PCRc y RT-qPCR, respectivamente.
Ensayos de Especificidad
Para estimar la especificidad del método se emplearon varios genomas virales y bacterianos patológicamente relacionados: IBV, SHS, ILTV, Avibacterium paragallinarum, Ornithobacterium rhinotracheale y Gallibacterium anatis. Se empleó un fragmento amplificado para NDV LaSota en el RT-PCR y un pico específico de disociación para el RT-qPCR (Jennings et al., 2009; Berwouts et al., 2010; Mattocks et al., 2010). Los ensayos se realizaron por triplicado.
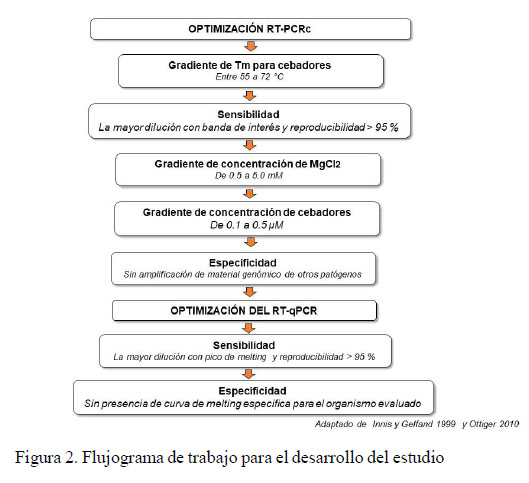
El flujograma de actividades implementadas en el estudio se muestra en la Figura 2.
RESULTADOS
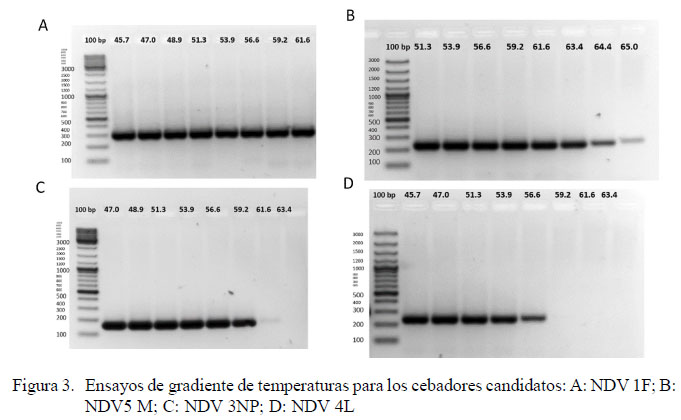
Una de las primeras fases para el proceso de estandarización de métodos moleculares (Figura 2) de RT-PCR fue determinar la temperatura ideal en la cual los cebadores hibridan con la secuencia de nucleótidos target, para lo cual se realizó una gradiente de temperaturas. En la Figura 1 se observan los resultados obtenidos por ensayos empíricos para los cuatro cebadores candidatos, donde los cebadores NDV 1F y NDV 5M presentan un amplio rango de temperaturas (45.7 a 61.6 °C y 51.3 a 63.4 °C, respectivamente), con la banda de interés definida y sin presencia de productos inespecíficos de alto o bajo peso molecular. El hecho de que puedan hibridar a temperaturas elevadas disminuye la presencia de bandas inespecíficas, mientras que los cebadores NDV 3NP y NDV4L presentaron un estrecho rango de temperaturas a elegir (47 a 59.2 °C y 45.7 a 56.6 °C, respectivamente), donde las temperaturas más bajas podrían propiciar la formación de inespecificidades, pese a que las bandas del producto de interés fueron definidas (Figura 3). Las temperaturas de hibridación optimizadas fueron 59.2, 56.6, 53.9 y 61.6 °C para los cebadores NDV 1F, NDV 3 NP, NDV 4L y NDV 5M, respectivamente.
En los ensayos de sensibilidad se determinó la cantidad del genoma de NDV, en su equivalente de cADN, que podía ser detectado con cada par de cebadores. En la Figura 4 se aprecia que los cebadores NDV1F, NDV5M y NDV4L son capaces de detectar hasta 10 fg de cADN, mientras que los cebadores NDV 3NP son 10 veces menos sensibles. Si bien es cierto que tres pares de cebadores tuvieron el mismo nivel de sensibilidad, los cebadores NDV 5M pudieron hibridar a una temperatura superior a 60 °C, lo que mejoraría la discriminación del fragmento de interés y reduciría la formación de dímeros entre cebadores, por lo que se consideraron para la optimización final.
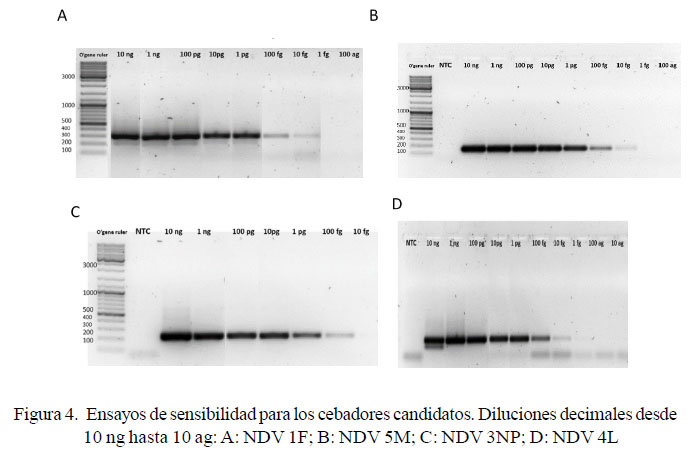
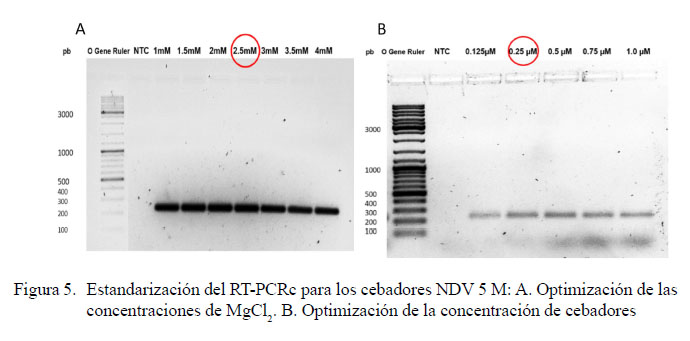
Para la estandarización del RT-PCRc en dos pasos se optimizó la concentración de MgCl2 y de los cebadores NDV 5M. Con respecto al MgCl2 se puede notar que la concentración ideal fue de 2.5 mM (Figura 5A), mientras que para la concentración de cebadores fue 0.25 µM (Figura 5B).
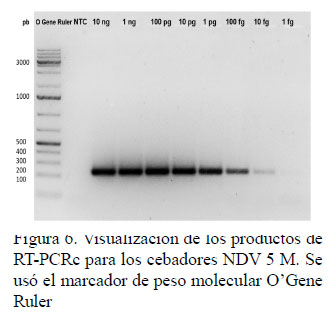
Con las concentraciones de los componentes de la reacción de RT-PCR convencional optimizada se llevó a cabo un segundo ensayo de sensibilidad en donde se pueden apreciar el producto de PCR de interés desde 10 ng hasta 1 fg, sin amplificados inespecíficos de alto o bajo peso molecular (Figura 6), por lo que el protocolo final tuvo las siguientes condiciones: denaturación inicial a 94 °C por 5 min, 35 ciclos de 90 °C por 30 s,45-65 °C por30 s,72 °C por 30 syun paso de extensión final a 72 °C por 5 min.
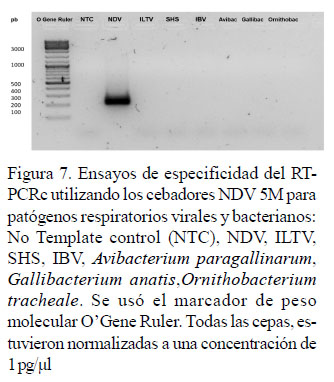
La especificidad depende de la secuencia de los cebadores. Por tal motivo, en los ensayos de especificidad para el RT-PCRc se observó que no hubo amplificación para el material genómico de otros virus y bacterias respiratorias, demostrando que los cebadores Figura 6. Visualización de los productos de RT-PCRc para los cebadores NDV 5 M. Se usó el marcador de peso molecular O’Gene Ruler
NVD 5M son adecuados por no brindar reacciones cruzadas frente a material genómico de otros patógenos aviares (Figura 7).
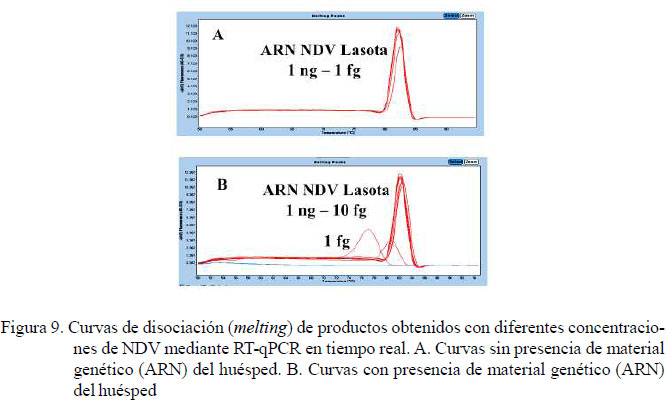
Luego de optimizar la plataforma de detección por RT-PCRc en dos pasos, se llevó a cabo los ensayos de RT-qPCR en un paso, cuyo programa consistió en 5 min de retrotranscripción reversa, 3 min para la activación de la enzima hot-start y 40 ciclos de 95 °C por 10 s, hibridación de 60 °C por 20 s y extensión de 72 °C por 20 s. Se observa que el RT-qPCR tuvo una sensibilidad de hasta 1 fg/µl, que pudo ser discriminada en base a la curva de disociación (Figura 8). Esto demuestra que la técnica de RT-qPCR es 10 veces más sensible que el método convencional; sin embargo, al introducir material genético del huésped se reduce la sensibilidad hasta 10 veces (Figura 9).
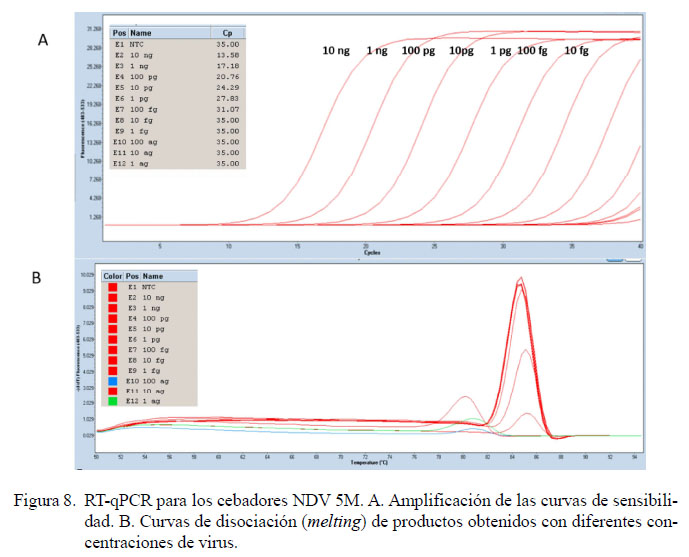
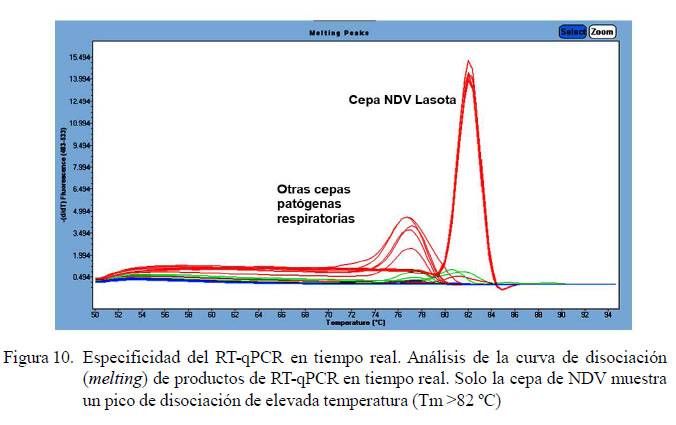
Los ensayos de especificidad para el RTqPCR demostraron presencia de un pico de interés con una temperatura mayor a 82 °C a diferencia de los picos generados por otras cepas patógenas respiratorias (72-80 °C) (Figura 10). El uso de agentes fluorescente como el Sybr Green I exige el uso de estas curvas para la confirmación de los verdaderos positivos. Esta técnica es sencilla de realizar y menos costosa en comparación con las sondas fluorescentes.
DISCUSIÓN
En este estudio se ha desarrollado una plataforma específica y sensible de RT-qPCR para la detección y cuantificación de NDV producido en un sistema de huevos embrionados SPF. La búsqueda bibliográfica dio como resultado la presencia de al menos cuatro pares de cebadores para detectar NDV de clase I (Farkas et al., 2009; Fuller et al., 2010). Estos cebadores fueron sometidos a una evaluación para determinar el más apropiado para la detección de NDV en muestras de fluido alantoideo. Se comparó la temperatura de hibridación óptima sugerida por el fabricante con la temperatura óptima determinada por análisis empírico (Rochelle et al., 1997), indicando los resultados que la temperatura optimizada fue mayor (56.6 y 61.6 °C) para los cebadores NDV 3NP y NDV 5M y menor (59.2 y 53.9 °C) en los cebadores NDV 1F y NDV 4L, difiriendo con la calculada por el fabricante. Resultados similares fueron encontrados por Rochelle et al. (1997) al evaluar seis pares de cebadores para la detección de Cryptosporidium parvum y Giardia lamblia.
Los cebadores NDV 5M obtuvieron los mejores resultados, debido a que las temperaturas de hibridación elevadas mejoran la discriminación entre dímeros de cebadores y extensión con nucleótidos erróneos en el extremo 3´ (Figura 3B). Esta característica y su capacidad para detectar hasta 10 fg de cDNA de NDV le permitieron continuar las siguientes etapas de optimización que incluyeron la gradiente de concentración de MgCl2 y cebadores, determinándose que la concentración de ambos componentes se encontraba dentro de los rangos establecidos (Innis y Gelfand, 1999).
Para incluir a los cebadores NDV5M en el desarrollo de la plataforma de RT-qPCR en un paso, se optimizó en el protocolo de PCR el tiempo de extensión, de 1 a 20 s, obteniendo mejores resultados con respecto a lo indicado por el fabricante (datos no mostrados). Luego se determinaron la sensibilidad y especificidad. Acorde a la literatura, los cebadores elegidos NDV 5M han demostrado ser uno de los más conservados (Cattoli et al., 2011) en comparación con las otras secuencias. Los cebadores NDV3P (Tan et al., 2004) fueron diseñados para la proteína F, obteniendo una baja sensibilidad (desde 10 ng hasta 10 pg/µl). Tan et al. (2009) utilizaron los NDV3P, asumiendo que la proteína NP es más conservada que la proteína F; sin embargo, su ensayo no mostró alta sensibilidad (109 a 105 copias de DNA plasmídico/ reacción) por lo que no formaron parte del protocolo final. Los cebadores NDV 4L, diseñados por Fuller et al. (2010) tuvieron alta sensibilidad, pero mostraron inespecificidades.
El uso de ssARN genómico purificado en lugar de plásmidos reflejan las condiciones naturales en las cuales es hallado el virus, lo cual es una ventaja en la determinación de la sensibilidad. Por otro lado, los cebadores de NDV 1F diseñados por Jang et al. (2011) presentaron bandas de interés menos definidas y su sensibilidad solo fue de aproximadamente de 400 copias virales/µl. Si bien es cierto, estos autores desarrollaron un protocolo de RT-qPCR para la detección y cuantificación de la cepa vacunal NDV LaSota producida en un sistema de biorreactores, se ha demostrado que este tipo de sistemas genera, por lo general, menores títulos virales que los alcanzados en huevos SPF, además de ser económicamente menos accesible (MacGuines et al., 2006). A su vez, los cebadores NDV5M diseñados por Seal et al. (1995), orientados hacia la proteína matriz, tuvieron como objetivo caracterizar cepas de NDV de campo por secuenciación directa y filogenia por lo cual su sensibilidad no fue determinada. En este estudio se determinó su sensibilidad para la cepa vacunal NDV LaSota, llegando a detectar hasta 116 copia virales/µl (1 fg/µl) por RTqPCR, demostrando que el gen M es una de los más conservados (Cattoli et al., 2011).
Los resultados de especificidad demostraron que no hubo amplificación para virus y bacterias patogénicamente relacionados. El desarrollo de esta plataforma de cuantificación por técnicas moleculares con condiciones optimizadas se puede emplear en el proceso de monitoreo de carga viral en la producción de vacunas de NDV LaSota.
CONCLUSIONES
Se desarrolló una plataforma molecular en base al gen matriz (M) para la cuantificación NDV a partir de un sistema de cultivo en huevos embrionados SPF. Se optimizaron los parámetros del RT-PCRc y se continuó con el desarrollo de RT-qPCR, el cual fue 10 veces más sensible que el RT-PCRc.
LITERATURA CITADA
1. Alexander DJ, Aldous EW, Fuller CM. 2012. The long view: a selective review of 40 years of Newcastle disease research. Avian Pathol 41: 329-335. doi: 10.1080/03079457.2012.697991 [ Links ]
2. Berwouts S, Morris MA, Dequeker E. 2010. Approaches to quality management and accreditation in a genetic testing laboratory. Eur J Hum Genet 18(Suppl 1): S1-S19. doi: 10.1038/ ejhg.2010.104 [ Links ]
3. Cattoli G, Susta L, Terregino C, Brown C. 2011. Newcastle disease: a review of field recognition and current methods of laboratory detection. J Vet Diagn Invest 23: 637-656. doi: 10.1177/ 1040638711407887 [ Links ]
4. Farkas T, Szekely E, Belak S, Kiss I. 2009. Real-time PCR-based pathotyping of Newcastle disease virus by use of TaqMan minor groove binder probes. J Clin Microbiol 47: 2114-2123. [ Links ]
5. Fuller CM, Brodd L, Irvine RM, Alexander DJ, Aldous EW. 2010. Development of an L gene real-time reverse-transcription PCR assay for the detection of avian paramyxovirus type 1 RNA in clinical samples. Arch Virol 155: 817-823. doi: 10.1007/s00705-010-0632-1 [ Links ]
6. Gallili GE, Ben-Nathan D. 1998. Newcastle disease vaccines. Biotechnol Adv 16: 343-366. doi: 10.1016/S0734- 9750(97)00081-5 [ Links ]
7. Innis M, Gelfand DH. 1999. Optimization of PCR: conversations between Michael and David. PCR Applic 1999: 3-22. doi: 10.1016/B978- 012372185-3/50002-X [ Links ]
8. Jang J, Hong SH, Kim IH. 2011. Validation of a real-time RT-PCR method to quantify Newcastle disease virus (NDV) titer and comparison with other quantifiable methods. J Microbiol Biotechnol 21: 100-108. doi:10.4014/ jmb.1006.06006 [ Links ]
9. Jennings L, Van Deerlin VM, Gulley ML. 2009. Recommended principles and practices for validating clinical molecular pathology tests. Arch Pathol Lab Med 133: 743-755. doi: 10.1043/ 1543-2165-133.5.743 [ Links ]
10. Ke GM, Cheng HL, Ke LY, Ji WT, Chulu JL, Liao MH, Chang TJ, Liu HJ. 2006. Development of a quantitative Light Cycler real-time RTPCR for detection of avian reovirus. J Virol Methods 133: 6-13. doi: 10.1016/ j.jviromet.2005.09.011 [ Links ]
11. McGinnes LW, Pantua H, Reitter J, Morrison TG. 2006. Newcastle disease virus: propagation, quantification, and storage. Curr Protoc Microbiol 15: 15F.2. doi: 10.1002/9780471729259.mc15f0- 2s01 [ Links ]
12. Mattocks CJ, Morris MA, Matthijs G, Swinnen E, Corveleyn A, Dequeker E, et al. 2010. A standardized framework for the validation and verification of clinical molecular genetic tests. Eur J Hum Genet 18: 1276-1288. doi: 10.1038/ ejhg.2010.101 [ Links ]
13. [OIE] World Organization for Animal Health. 2017. Manual of diagnostic tests and vaccines for terrestrial animals. Ch. 2.3.14. Newcastle disease (infection with Newcastle disease virus). Paris. [Internet]. Available in: http:// www.oie.int/fileadmin/Home/eng/ H e a l t h _ s t a n d a r d s / t a h m / 2.03.14_NEWCASTLE_DIS.pdf [ Links ]
14. Ottiger HP. 2010. Development, standardization and assessment of PCR systems for purity testing of avian viral vaccines. Biologicals 38: 381-388. doi: 10.1016/j.biologicals.2010.01.015 [ Links ]
15. Rochelle PA, De Leon R, Stewart MH, Wolfe RL. 1997. Comparison of primers and optimization of PCR conditions for detection of Cryptosporidium parvum and Giardia lamblia in water. App Environ Microbiol 63: 106-114. [ Links ]
16. Roux KH. 2009. Optimization and troubleshooting in PCR. Cold Spring Harb Protoc 2009(4):pdb.ip66. doi: 10.1101/ pdb.ip66 [ Links ]
17. Seal BS, King DJ, Bennett JD. 1995. Characterization of Newcastle disease virus isolates by reverse transcription PCR coupled to direct nucleotide sequencing and development of sequence database for pathotype prediction and molecular epidemiological analysis. J Clin Microbiol 33: 2624-2630. [ Links ]
18. Senne DA, King DJ, Kapczynski DR. 2003. Control of Newcastle disease by vaccination. Dev Biol (Basel) 119: 165-170. [ Links ]
19. Tan SW, Omar AR, Ideris A, Yusoff K, Tan WS. 2004. Detection of Newcastle disease virus using a SYBR Green I real time polymerase chain reaction. Acta Virol 48: 23-28. [ Links ]
20. Tan SW, Ideris A, Omar AR, Yusoff K, Hair-Bejo M. 2009. Detection and differentiation of velogenic and lentogenic Newcastle disease viruses using SYBR Green I real-time PCR with nucleocapsid gene-specific primers. J Virol Methods 160: 149-156. doi: 10.1016/ j.jviromet.2009.05.006 [ Links ]
Recibido: 28 de octubre de 2017
Aceptado para publicación: 3 de abril de 2018

