











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO  uBio
uBio 
 Permalink
PermalinkINTRODUCCIÓN
Para el burro o asno doméstico (Equus asinus) se han desarrollado varias teorías sobre sus orígenes. Un planteamiento consideró que desciende del onager asiático (Equus hemionus), del asno de Nubia (Equus asinus africanus) como ancestro de la raza Andaluza, o del asno somalí (Equus asinus somaliensis) que dio origen a los burros del sudoeste de Asia y algunas razas europeas, así como del Equus asinus europeus cuya área de origen es la cuenca mediterránea, dando origen a la mayoría de los burros europeos (Aranguren-Méndez et al., 2001; Beja- Pereira et al., 2004; Kimura et al., 2011).
El burro fue introducido al continente americano en el siglo XVI, y durante la colonia se distribuyó en todo México, asociado a labores agrícolas, de carga y transporte. Por sus orígenes es considerado como criollo, y es parte de la cultura y costumbres de ciertas regiones del país; no obstante, la cría y explotación de los híbridos (mulas y burdéganos) tuvo un impacto similar o de mayor trascendencia (Álvarez-Romero et al., 2008). Las poblaciones ferales descienden de burros domésticos liberados o escapados y se localizan en varias zonas fuera de su área de distribución original (García, 1994; Álvarez- Romero et al., 2008). La Asociación Mexicana de Criadores de Burros y Mulas (AMCBM) se constituyó con el objetivo de promover la cría, conservación y mejoramiento genético del burro criollo mexicano (BCM), al cual denominaron Burro Mixteco, como una raza de origen mexicano (AMCBM, 2018).
La diversidad genética es la base para satisfacer las necesidades de producción en diversos entornos, programas de conservación, esquemas de mejoramiento genético sostenible y adaptación de las poblaciones a los cambios en los objetivos de producción a través de ambientes (Alderson, 2018). El manejo efectivo de los recursos genéticos y el análisis de los componentes de diversidad genética son parte de las áreas estratégicas prioritarias del plan global de acción para los recursos zoogenéticos, y que fueron definidas en la Conferencia Técnica Internacional sobre los Recursos Zoogenéticos para la Agricultura y la Alimentación, realizada en Interlaken, Suiza y promovida por la FAO (FAO, 2007). El objetivo del presente estudio fue evaluar la diversidad genética en BCM con marcadores genéticos de tipo SNP (Polimorfismo de Nucleótido Único). El estudio proporciona las bases para definir el panel de marcadores genéticos a utilizar en la caracterización, pruebas genéticas y desarrollo de la población del BCM.
MATERIALES Y MÉTODOS
Se muestreó a 16 individuos (ocho machos y ocho hembras) susceptibles de registro en la AMCBM sin verificar niveles de parentesco. Las muestras de sangre fueron remitidas al laboratorio de Neogen GeneSeek (USA). Los genotipos analizados conforman el chip GGP Equine 70K (71 947 loci). Dentro de los cromosomas (30 autosómicos y uno sexual), se identificaron los SNP polimórficos donde la frecuencia del alelo menor (FAM) fue igual o mayor a 0.01. Con base a las frecuencias alélicas se estimó la heterocigosis esperada (He) y observada (Ho), contenido de información polimórfica (CIP) y el índice de fijación (FIS) y equilibrio (HW) Hardy- Weinberg (Nei y Chesser, 1983; Nei, 1987; Weir, 1996). Los niveles de He, Ho y CI determinan si un marcador genético es o no informativo y su potencial de uso en estudios de variabilidad genética. El estadístico FIS mide la reducción de heterocigosis y es una medida del grado de endogamia en una población. El posible desequilibrio de ligamiento (DL) se evaluó con base en la correlación (r2) entre frecuencias a través de locus (Waples, 2006). El r2 fluctúa de cero a uno, donde valores alrededor de cero indican ausencia de DL y segregación independiente y conforme aumenten hacia la unidad es suposición de asociación no aleatoria entre locus.
Para identificar componentes de varianza entre grupos y dentro de grupos, se realizó un análisis de varianza molecular (AMOVA). Los análisis genéticos y estadísticos se realizaron con los programas GenAlex 6.051 (Peakall y Smouse, 2012) y FSTAT (Goudet, 1995). Para determinar la posible estructura dentro de la población se realizó el estudio de agrupamiento bayesiano con el software Structure (Pritchard et al., 2000) para inferir el número de genotipos o clusters (k) apropiados. Se evaluaron cinco niveles a partir del análisis de genotipos utilizando múltiples loci (k=1 a k=5) y los individuos fueron asignado probabilísticamente a los grupos (k). La selección del k ajustado se realizó con base al valor óptimo descrito por Evanno et al. (2005), calculado con el software online Structure Harvester (Earl y vonHolt, 2012).
RESULTADOS
De los SNP evaluados, 3579 loci (4.9%) presentaron variabilidad genética donde la FAM fue superior a 0.29; además, el 24.9% presentó desequilibrio HW (p<0.05). Según el cromosoma (Cuadro 1), el número de loci polimórficos osciló de 50 a 269 con un valor promedio de 115. Los valores promedio (dentro de cromosoma) para CIP y FIS fluctuaron entre 0.222 y 0.267 y de -0.392 a -0.208, respectivamente. En todos los cromosomas el valor de Ho fue superior a He y el signo negativo de FIS señala gran cantidad de heterocigotos.
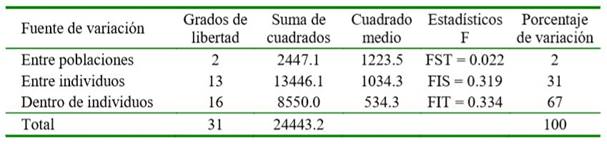
Con relación al DL, los valores promedio de r2 dentro de cromosoma fueron superior a 0.10; no obstante, se observan r2 superiores a 0.50, lo cual señala que algunos loci presentan DL y deben de ser analizados a detalle. En la Figura 1 se presentan los resultados para el análisis de estructura de la población, donde el K óptimo correspondió a 2, con una contribución genética promedio de 63.1% y 36.9%. En las imágenes, cada columna pertenece a un individuo evaluado y la longitud de los distintos colores definen la proporción de los genomas o cluster identificados. El análisis de varianza molecular (Cuadro 2) expuso que la variabilidad genética dentro de individuos explicó el 67% de la varianza genética total.
Cuadro 1 Número de SNP polimórficos e indicadores de variabilidad genética a través de cromosomas
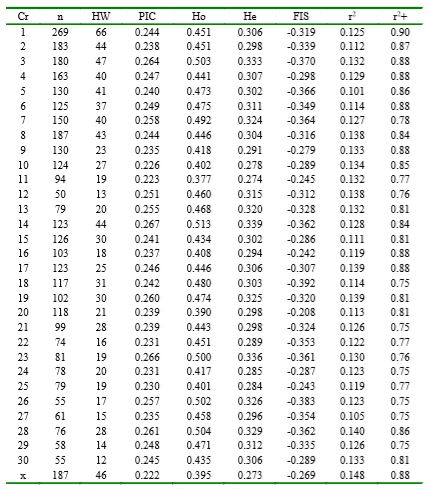
Cr, cromosoma; N, número de loci polimórficos; HW, número de loci que presentaron desequilibrio (p<0.05) Hardy Weinberg; PIC, valor promedio del contenido de información polimórfica; Ho, valor promedio de la heterocigosis observada; He, valor promedio de la heterocigosis esperada; FIS, valor promedio del estadístico FIS; r2, valor promedio de la correlación entre frecuencias a través de locus; r2+, valor máximo de las correlaciones entre frecuencias a través de locus
DISCUSIÓN
Se han reportado resultados diversos para la eficiencia en el uso de chips, a través de razas y de especies. McCue et al. (2012) con base en el Equine SNP 50k determinó en 14 razas de caballos que el porcentaje de SNP con información polimórfica fluctúo de 68 a 87%; no obstante, en 18 especies del género Equus el porcentaje de locus con al menos un heterocigoto fluctuó de 0.518 a 1.56.
Con el objetivo de diseñar un panel para pruebas genéticas en burros, se identificaron los 101 SNP propuestos por Holl et al. (2017), de los cuales solo cuatro presentaron CIP. Con relación a estudios sobre el origen y domesticación del asno, Rosenbom et al. (2015) utilizando un panel de microsatélites analizaron poblaciones de ocho países del noreste de África y Asia occidental, señalando como principal población de origen al asno del noreste de África (Equus africanus africanus); sin embargo, identificaron una fuente de variación genética alterna en la población de Yemen, que puede ser asociada a la domesticación del asno. Beja-Pereira et al. (2004) con ADN mitocondrial y muestras de 52 países identifican dos grupos divergentes en el árbol filogenético señalando como ancestros al Equus hemionus y Equus kiang para un grupo, así como el Equus asinus somaliensis y el Equus africanus para otro grupo. Por otro lado, Kimura et al. (2011) describen las aportaciones del Equus africanus africanus y el Equus africanus somaliensis al proceso de domesticación y divergencia genética de las poblaciones actuales.
Las poblaciones de ganado criollo en América Latina procedentes de la península Ibérica han evolucionado en ambientes diversos y adversos, lo que sugiere que poseen genes para adaptación en frecuencias distintas a las poblaciones de origen (Núñez- Domínguez et al. 2016). Jordana et al. (2012) evaluando poblaciones de 11 países de América y razas europeas de asnos, reportaron que la estructura de la población fue definida por dos cluster (k=2). Asimismo, indican que el limitado flujo de animales entre Europa y América ha permitido una progresiva diferenciación genética de las poblaciones asnales americanas; incluso una separación entre poblaciones de centro y sur América. El BCM está en el marco de las poblaciones criollas procedentes de la península Ibérica, donde el análisis de estructura de población reveló que la variabilidad genética es producto de dos genotipos o cluster (Figura 1). En la península Ibérica se encuentran definidas cinco razas: Andaluza, Catalán, Encartaciones, Zamorano y Mallorquina (Rodero et al., 1998)). Al respecto, Aranguren-Méndez et al. (2001) con microsatélites desarrollados para pruebas genéticas en equinos evaluaron la diversidad genética de estas razas como ancestrales del BCM, reportando niveles para Ho y CIP de 0.528 a 0.570 y de 0.20 a 0.80, respectivamente.
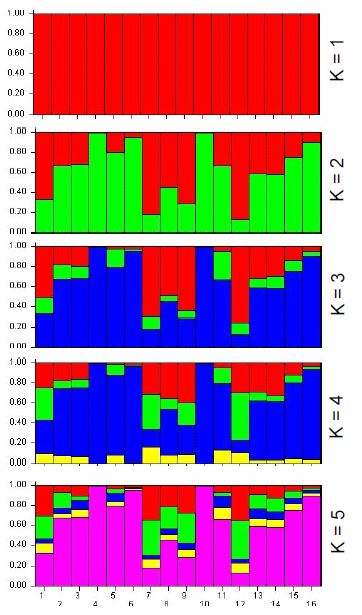
Figura 1 Resultados para la estructura genética de la población de 16 burros analizada (K, número de cluster evaluados) con el software Structure (Pritchard et al., 2000)
En otros estudios afines, Ivankovic et al. (2002) en tres razas asnales de Croacia, señalan que el 2.7% de la variabilidad se atribuyó a diferencias entre poblaciones y el 97% a diferencias dentro de individuos. Yun y Cho (2017) en asnos nativos de Corea del Sur reportaron niveles de Ho y CIP en los intervalos de 0.20 a 0.88 y de 0.22 a 0.89, respectivamente, en tanto que Zeng et al. (2019) en 12 razas de asnos en China, reportaron que la estructura de la población fue conformada por dos líneas o cluster, además los resultados para Ho y FIS oscilaron de 0.54 a 0.59 y de 0.03 a 0.14, respectivamente.
CONCLUSIONES
Los SNP identificados como polimórficos, conforman un primer panel de marcadores genéticos para el burro criollo mexicano; con aplicaciones para próximos estudios relativos a genómica (identidad racial, paternidad, distancias genéticas, huellas de selección, entre otros). Por otro lado, la variabilidad genética estimada puede ser utilizada en esquemas de desarrollo y conservación de la población.

