










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
Print version ISSN 1726-4634
Rev. perú. med. exp. salud publica vol.18 no.1-2 Lima Jan./jun. 2001
TRABAJOS ORIGINALES
Síntesis in vitro de la proteína de la envoltura del virus peruano de la fiebre amarilla
Carlos Yábar V1; Ysabel Montoya P1.
1 División de Biología Molecular, Centro Nacional de Salud Pública, Instituto Nacional de Salud.
RESUMEN
Objetivo: Sintetizar la proteína recombinante de la envoltura (Er) del virus peruano de la Fiebre Amarilla (FA) utilizando técnicas moleculares. Materiales y métodos: El gen de la proteína de interés fue amplificado por transcripción reversa - reacción en cadena de la polimerasa (RT-PCR) y clonado en un vector plasmídico para ser analizado mediante secuenciamiento de ADN. El inserto de ADN fue subdonado en un vector de expresión para ser traducido a proteína. Se purificó la proteína mediante cromatografía de afinidad bajo condiciones denaturantes, siendo visualizada por electroforesis en SDS-PAGE. Resultados: Se sintetizó y purificó la pr E del virus de la FA. Presentó un peso de 66 kDa, y luego de tres horas de inducción a partir de 1x108 células (OD=0.5) se obtuvo 10 mg/mL de la proteína a miniescala. Conclusión: El análisis de la secuencia de aminoácidos demostró que Er podría ser un buen candidato a ser evaluado serológicamente y luego ser usado como herramienta de diagnóstico específico para la FA.
Palabras clave: Fiebre amarilla; Proteína / aislamiento & purificación; Reacción en cadena por polimerasa de transcriptasa reversa (fuente: BIREME).
ABSTRACT
Objective: To synthesise envelope recombinant protein (Er) from Peruvian yellow fever virus (FA) by using molecular approaches. Materials and Methods: The gene that codes this protein was analysed by RT-PCR, then cloned into a plasmid vector and sequenced by using DNA sequencing. Later, the DNA insert was subdoned into an expression vector to be translated to protein. Results: The protein was purified through an affinity column under denaturing conditions and visualised by SDS-PAGE electrophoresis. Results: The recombinant protein was 66 kDa molecular weight and it was obtained about 10 mg/ml from 1x108 cell (OD=0.5) after three hours of induction. Conclusion: Amino acid sequence analysis showed that Er might be a good candidate to be tested using sera and then to be used as a specific yellow fever diagnosis tool.
Keys words: Yellow fever; Protein/isolation & purification; Reverse transcriptase polymerase chain reaction (source: BIREME).
INTRODUCCIÓN
La Fiebre amarilla (FA) es una enfermeclad viral infecciosa que afecta a cerca de 200,000 personas al año en todo el mundo y se estima que ha causado 30,000 muertes1.
En el Perú esta enfermedad ha cobrado un alto número de víctimas en los útimos decenios2 y, pese a que existe una vacuna eficaz para controlar esta enfermedad, la mortalidad y morbilidad en nuestro país sigue sigue siendo un problema de salud. Un inconveniente es la dificultad de diferenciar la FA de otros síndromes icterohemorrágicos como malaria, bartonelosis, leptospirosis y hepatitis, cuyos síntomas clínicos suelen confundirse3. Por ello, se recurre a las pruebas de diagnóstico serológico como ELISA de captura de IgM e IgG (MAC-ELISA y GAC-ELISA, respectivamente)4, pruebas inmunológicas altamente sensibles y de bajo costo, usadas como rutina en nuestro país. Sin embargo, su especificidad es baja debido al alto índice de reactividad cruzada con otros flavivirus relacionados, como el virus dengue y, posiblemente, el virus de la encefalitis de San Luis, entre otros.
La reactividad cruzada se debe a la estrechez genética entre el grupo de los flavivirus, cuyos epítopes presentes en sus antígenos pueden ser reconocidos por anticuerpos que presentan reactividad frente a otros virus del mismo grupo5. Este problema dificulta la confirmación diagnóstica, puesto que las pruebas serológicas utilizan antígenos totales del virus de la FA (cóctel de proteínas que incluyen antígenos altamente conservados entre los flavivirus).
Por ello, sería importante utilizar un solo antígeno del virus de la FA altamente antigénico. Un candidato a ser usado como herramienta diagnóstica es la glicoproteína de la envoltura (gp E), primer antígeno viral reconocido por los anticuerpos neutralizantes del hospedero durante el proceso de infección6 y que además presenta propiedades inmunogénicas en ratones7.
En ese sentido muchos investigadores han aprovechado las características de la proteína E para analizar la presencia de epítopes de reactividad cruzada y específicos entre grupos de flavivirus. De acuerdo a ello, la proteína de la envoltura del virus dengue presenta tres principales dominios; epitópicos denominados A, B y C. El dominio A comprende los aminoácidos de las posiciones 50-130 y 185-300 8, el dominio B se localiza entre los aminodcidos 298-400 9 y el dominio C está situado entre las posiciones 130-185 8. Las evidencias descritas en estos estudios demuestran la existencia de diferentes epítopes de valor diagnóstico en la proteína E del virus de la FA, que podría ser aprovechado en futuros ensayos serológicos.
El presente trabajo reporta la síntesis y purificación in vitro de la proteína recombinante E del virus de la FA utilizando técnicas moleculares.
MATERIALES Y MÉTODOS
AISLAMIENTO VIRAL
Se trabajó con una cepa del virus de la FA obtenida de un paciente infectado procedente del departamento de San Martín (año 1999). La cepa viral fue cultivada en células C6/36 de mosquito Aedes albopictus y mantenida en un medio enriquecido MEM.
EXTRACCIÓN DE ARN
El ARN total fue extraído a partir de la muestra de cultivo usando un kit de extracción de ARN QlAmp viral RNA kit (QIAGEN) de acuerdo a las recomenclaciones de sus fabricantes 10. Para tal efecto, un volumen de 140 ml de cultivo viral fueron lisados utilizando una solución denominada AVL, añadiéndose la mezcla total a una columna de purificación, se centrifugó a 8000 RPM por un minuto y las impurezas remanentes fueron removidas mediante sucesivos lavados con etanol. Nuevamente se realiz6 una centrifugaci6n a 8000 RPM por un minuto, siendo el ARN recuperado en tubos de 1.5 mL de capacidad mediante elusión con agua libre de ARNasas y almacenados a –80°C.
TRANSCRIPCIÓN REVERSA-REACCIÓN EN CADENA DE LA POLIMERASA
40 ng de ARN fue sometida a una reacción de transcripción reversa en solución conteniendo 200 U de la enzima transcriptasa reversa Supercript II, 25 mM Tris-HCl, pH 8,3, 37,5 mM KCl, 1,5 mM MgCl2 y 1 mM DTT (Gibco), 200mM de 0,2 mM de dATP, dCTP, dGTP y dTTP y finalmente 20 nmoles de primer reverso YF2 11. La mezcla de reacción fue incubada a 42°C por 60 minutos.
Para realizar la reacción en cadena de la polimerasa (PCR), 400 pg de ADN complementario (ADNc) fueron mezclados en un tubo conteniendo 10 mM Tris-HCl, pH 8,3, 50 mM KCl, 1.5 mM MgCl2 0.01 % en w/v de gelatina, 2 mM Cl2Mg, 0.2 mM de cada uno de los cuatro nucleótidos trifosfatos y 20 nmoles del primer reverso YF2 y el primer forward YF711, y 0,05U/ml de Taq ADN polimerasa. La muestra fue procesada en un termociclador GeneAmp PCR System 9600 (Perkin-Elmer) por 35 ciclos. Cada ciclo consistió de 95°C por 30 seg, 56°C por 90 seg y 72°C por 2 min con una extensión final de 72°C por 10 min. Para visualizar las bandas de ADN, una alícuota de 5 mL del producto final fue resuelto en un gel de agarosa 1%. Posteriormente, el gel fue teñido con bromuro de etidio y las bandas visualizadas mediante fluorescencia con rayos ultravioleta.
CLONAMIENTO Y SECUENCIAMIENTO
El producto de amplificación fue purificado y ligado en un vector plasmídico pGEM-T easy (Promega). El plásmido recombinante denominado pGEM-FA99 fue introducido en bacterias E coli. cepa XL1 Blue mediante electroporación. Las colonias recombinantes fueron rastreadas mediante un ensayo de minipreparación de plásmidos. Los plásmidos que portaron el inserto de ADN de interés fueron purificados y secuenciados mediante el método de terminación de dideoxinucleótidos usando un secuenciador automático ALF Express (Pharmacia).
SUBCLONAMIENTO Y SÍNTESIS DE LA PROTEÍNA RECOMBINANTE
Una concentración de 5 ng del plásmido pGEM-FA99 fue sometido a PCR usando primers modificados con sitios de restricción YF7BgII y YF2HindIII. El producto resultante denominado FA99 fue digerido con 10 U de las enzimas de restricción BgII y HindIII. Al mismo tiempo, el plásmido de expresión pQE40 (QIAGEN) fue seleccionado para llevar a cabo la subclonación del producto de amplificación FA99-BgII-HindII. Para tal efecto, 1 μg de pQE40 fue digerido con 10 U de las enzimas BgII y HindII. El inserto de ADN y el plásmido fueron ligados y el plásmido recombinante pQE40-FA99 fue utilizado para transformar bacterias XL1-BLUE. Las bacterias recombinantes fueron seleccionadas mediante preparación de ADN plasmídico.
La síntesis de la proteína recombinante se realizó siguiendo las recomendaciones de los fabricantes del Kit QIA expressionist 12 con algunas modificaciones: las bacterias portando la construcción pQE40-FA99 fueron incubadas en caldo LB ampicilina por toda la noche. Un inóculo de 1,5 ml de cultivo de toda la noche fue mezclado con 3 mL de caldo LB ampicilina fresco e incubado por tres horas hasta que el OD=0.5. Una concentración de 1 mM de isopropiltiogalactopiranósido (IPTG) fue inoculado para inducir la síntesis de la proteína. Las bacterias fueron recuperadas por centrifugación a 12000 RPM por 6 minutos y el pellet fue resuspendido en una solución de lisis A (6M de Guanidina hidroclorhidra, 0,1 M de NaH2PO4, 0.01 M de Trizma-HCl, pH 8.0). El lisado fue centrifugado a 14000 RPM por un minuto y el sobrenadante fue recuperado en un tubo nuevo. Esta solución fue mezclada con 50 mL de resina Ni-NTA e incubada por 30 minutos con movimiento constante. La resina fue precipitada por centrifugación y los contaminantes fueron removidos mediante una solución de lavado conteniendo 8 M de úrea, 0.1 M NaH2PO4, 0,01 M de Trizma-HCl, pH 6,3. La proteína fue recuperada en tubos nuevos mediante un proceso de elusión usando una solución conteniendo 8 M de úrea, 0.1 M NaH2PO4, 0,01 M de Trizma-HCl, pH 4,5. Para la visualización de la proteína recombinante se realizó una electroforesis en SDS-PAGE, en donde el gel fue teñido en azul brillante de Coomasie y las bandas fueron visualizadas mediante lavados con solución decolorante.
ANÁLISIS
Se emplearon para el análisis de alineamiento de secuencias e hidrofobicidad de la proteína E del virus de la FA dos paquetes software: Expasy http://ww.expasy.ch/ y Dialign 2,1 http://www.bibiserv.techjack.uni-bielefeld.de/dialing2/.
RESULTADOS
El inserto de ADN FA99 correspondió a una región genética que codifica una porción carboxiterminal de 337 aminoácidos de la proteína de la envoltura.
La secuencia de nucleótidos del ADNc del virus de la FA obtenida de la reacción de secuenciamiento denominada FA99, reveló un marco de lectura de 1011 pares de bases que codificaban 337 aminoácidos. Mediante el análisis de alineamiento con secuencias reportadas en el banco de genes, se pudo determinar que la región en estudio comprendió el 68% del gen entero, desde las posiciones 107 al 444 de los 493 aminoácidos totales. La zona en estudio correspondió a un porción carboxiterminal de la proteína. Cabe resaltar que la secuencia de aminoácidos de FA99 abarcó las regiones epitópicas de reconocimiento humoral (Ver Figura 1).
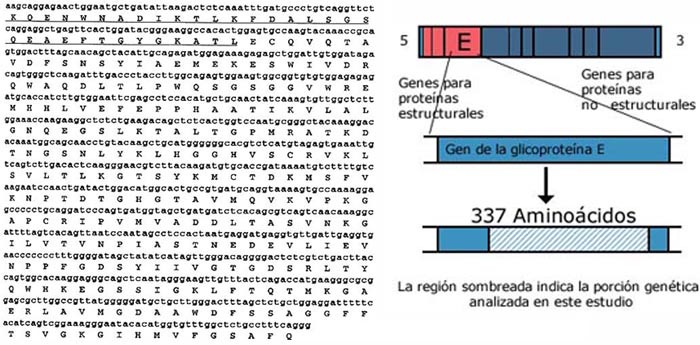
Figura 1. Secuencia de nucleótidos y de aminoácidos del gen
de la glicoproteína E del virus de la FA de San Martín año 1999.
Los epítodes o dominios antigénicos reportados por Roehrig8 y Simmons9
son señalados: la secuencia roja indica el dominio A, la secuencia subrayada
indica el dominio C y la secuencia verde, el dominio B.
El perfil hidropático de la secuencia de aminoácidos de FA99 fue conservado respecto a otras cepas de referencia de FA y Dengue.
Para determinar si la proteína de FA99 presentó un perfil primario similar a otros flavivirus se diseñó el perfil de hidropatía según Kyte (13), observándose diferentes picos hidrofóbicos e hidrofílicos altamente conservados entre una cepa y otra; sin embargo, la presencia de cambios aminoacídicos en FA99, trajo consigo la formación de nuevos picos hidrofílicos respecto a las otras cepas de referencia. La Figura 2 muestra que la proteína a sintetizarse podría presentar una característica altamente hidrofóbica, es decir, cargada negativamente.
De otro lado, es importante resaltar que esta región de la proteína presenta diferencias muy marcadas con otros flavivirus, especialmente con el virus dengue.
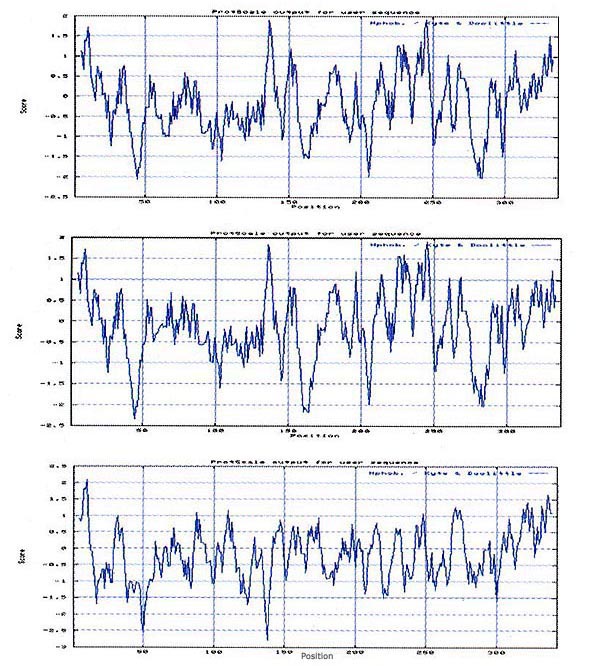
Figura 2. (A) Análisis de hidropatía de la secuencia de 337 aminoácidos
del gen de la glicoproteína E del virus de la FA, aislamiento de San Martín,
año 1999, Nº de acceso AF12554; (B) Aislamiento africano cepa 17D,
Nº de acceso X03700; (C) Virus Dengue 2, proveniente de Amazonas,
año 1996, Nº de acceso AF 093674.
Nota: El eje de las abcisas señala el índice de hidrofobicidad.
La proteína recombinante fue exitosamente sintetizada usando un sistema de expresión procarionte.
El análisis de SDS-PAGE mostró la presencia de una banda correspondiente a un tamaño de aproximadamente 66 kDa. Esta banda correspondió a una proteína de fusión conformada por la proteína de interés a partir de FA99 y una proteína de 26 kDa de la dehidrofolato reductasa de ratón (DHFR). La proteína fue obtenida en buena concentración usando sistemas denaturantes de purificación basado en úrea. Luego de tres horas de inducción a partir de 1x108 células (OD=0.5), pudo obtenerse aproximadamente 10 mg/mL de la proteína de interés a miniescala (Figura 3).
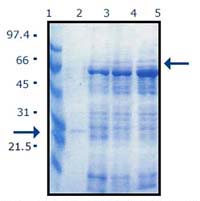
Figura 3. Electroforésis de SDS-PEG 10% para el análisis y visualización
de la proteína recombinante de la envoltura del virus de la FA, aislamiento peruano
(San Martín, 1999). 1º carril: Marcador estándar de peso molecular;
2º carril: Dehidrofolato reductasa de ratón; 3º, 4º y 5º carril:
Proteína de fusión recombinante inducida por 1, 2 y 3 horas,
respectivamente con isopropiltiogalactopiranosida (IPTG).
La flecha superior señala la proteína de fusión de la envoltura y la DHFR,
mientras que la flecha inferior señala la DFHR sola.
DISCUSIÓN
En el presente trabajo hemos seleccionado una porción genómica del gen de la proteína de la envoltura (E) del virus de la FA para ser sintetizada como proteína recombinante. Esta proteína es importante debido al rol que cumple durante los procesos de reconocimiento del virus al receptor Fc, y en la inducción de anticuerpos neutralizantes específicos 8. Estas características convierten a la proteína E en un antígeno alternativo en el diagnóstico específico de la FA en ensayos serológicos.
De otro lado, hemos realizado un análisis del perfil hidropático de la región estudiada sobre la base de su secuencia de aminoácidos. Este análisis es importante porque nos permite caracterizar picos hidrofílicos en la proteína. Cabe mencionar que la presencia de dominios con picos hidrofílicos podrían ser sitios potencialmente antigénicos, como fue demostrado previamente en el virus dengue 5. De acuerdo a nuestro análisis, es importante mencionar que FA99 presentó un perfil hidropático muy conservado con respecto a otras cepas del virus de la FA de referencia como la cepa Africana 17D. Sin embargo al comparar el perfil hidropático de FA99 con el de la proteína de la envoltura del virus dengue se observaron importantes diferencias, principalmente en algunos picos hidrofílicos (Figura 2). Esta observación es importante porque los anticuerpos inducidos por cada una de las proteínas, podrían presentar una especificidad preferencial por epítopes conformacionales. Este alcance podría ser aprovechado para la identificación de anticuerpos específicos contra el virus de la FA utilizando su propia proteína de envoltura.
Asimismo, ciertas zonas en donde se encuentran localizadas algunas regiones epitópicas no presentaron variación en su perfil hidropático, es decir, no hubo mayores cambios aminoacídicos de tipo no conservativos. La razón probablemente se deba a que ciertos dominios epitópicos de la proteína están involucrados en el reconocimiento del receptor Fc del macrófago. Estos dominios, por naturaleza, son altamente conservados entre los flavivirus, como es el caso del dominio B14.
Por otro lado, hemos logrado sintetizar la proteína recombinante usando técnicas moleculares bajo un sistema de expresión procarionte. El tamaño de la proteína visualizada en el gel de electroforesis SDS-PAGE corresponde al tamaño esperado de acuerdo a su secuencia de aminoácidos. La producción a miniescala de la proteína en E coli, incluyendo los procesos de extracción y purificación no demandan generalmente mucho esfuerzo ni material de trabajo.
Es importante resaltar que esta proteína podría ser una fuerte candidata a ser evaluada por serología y posteriormente ser utilizada como herramienta diagnóstica. De hecho, algunos autores han empleado construcciones de esta proteina recombinante para detectar anticuerpos frente al virus dengue en ensayos de ELISA15. Bajo este contexto, cabe mencionar que en la actualidad los sistemas de diagnóstico serológico convencionales no presentan una alta especificidad debido a la presencia de reactividad cruzada frente a otros flavivirus. En consecuencia, el uso de una sola proteína del virus, con menos epítopes de reactividad cruzada, podría mejorar esta inconveniencia de inespecificidad, principalmente en los sistemas MAC ELISA y GAC ELISA para el diagnóstico serológico de la FA. Asimismo, el uso de una proteína de fusión tipo DHFR disminuiría en gran magnitud los riesgos de reactividad inespecífica debido a la baja afinidad que existe entre los anticuerpos humanos y los epítopes de DHFR de ratón12.
Finalmente, es importante mencionar que otras proteínas candidatas a ser usadas como herramientas de diagnóstico están siendo sintetizadas. Entre ellas podemos mencionar a las proteínas C - prM, NS1 y NS3. Próximos experimentos usando ensayos de Western blot y ELISA permitirán seleccionar la proteína más antigénica y con menor reactividad cruzada o, en todo caso, usar un coctel de construcciones de fragmentos de proteínas que conserven los epítopes de mayor especificidad y antigenicidad.
AGRADECIMIENTOS
Al Consejo Nacional de Ciencia y Tecnología (CONCYTEC) por subvencionar parcialmente el presente proyecto, a la División de Virología del Instituto Nacional de Salud por proporcionamos la cepa viral de FA y a los Sres. Juana Choque Portilla y David Garcia Neyra, personal técnico del laboratorio de Biología Molecular del Instituto Nacional de Salud por su valiosa asistencia en las técnicas moleculares.
REFERENCIAS BIBLIOGRÁFICAS
1. Vainio J, Cutts F. Yellow Fever. Division of emergin and other communible diseases surveillance and control. Global programme for vaccines and inmunization expanded programme on inmunization. Geneva: OMS; 1998. [ Links ]
2. Carrillo C. Situación de la fiebre amarilla en el Perú. En: Libro de resúmenes de la reunión de expertos, estrategias de prevención y control de la fiebre amarilla. Riesgos de urbanización en las américas. Lima: INS/DISA Cusco/OPS; 1998. [ Links ]
3. Cabezas C. Manejo Clínico. En: Libro de resúmenes de la reunión de expertos, estrategias de prevención y control de la fiebre amarilla. Riesgos de urbanización en las américas. Lima: INS/DISA Cusco/OPS; 1998. [ Links ]
4. Corber S, Minaya P. Diagnóstico y vigilancia sindrómica. En: Libro de resúmenes de la reunión de expertos, estrategias de prevención y control de la fiebre amarilla. Riesgos de urbanización en las américas. Lima: INS/DISA Cusco/OPS; 1998. [ Links ]
5. Falconar AKI. Identification of an epitope on the dengue virus membrane (M) protein defined by cross-protective monoclonal antibodies: design of an pimproved epitope sequence based on common determinats present in both envelope (E and M) proteins. Arch Virol 1999; 144: 2313-30. [ Links ]
6. Ryman KID, Ledger TN, Weir FIC, Schlesinger JJ Barret DT. Yellow fever virus envelope protein has two discrete type-specific neutralizing epitopes. Journal of General Virology 1997; 78: 1353-6. [ Links ]
7. Kelly P, Greene JJ, Kimg AD, Innis B. Purified dengue 2 virus envelope glycoproteins aggegates produced by baculovirus are inmunogenic in mice. Vaccine 2000; 18: 2549-59. [ Links ]
8. Roehrig JT, Bolin RA, Kelly RG. Monoclonal antibody mapping of the envelope glycoprotein of the dengue 2 virus, Jamaica. Virology 1998; 246: 317-28. [ Links ]
9. Simmons M, Porter KR, Escamilla J, Graham R, Watts DM, Eckels KH, et al. Evaluation of recombinant dengue viral envelope B domain protein antigens for the detection of dengue complex-specific antibodies. Am J Trop Med Hyg 1998; 58(2): 144-51. [ Links ]
10. QIAGEN. QlAamp Viral RNA Mini Kit Handbook. For purification of viral RNA from Plasma, Serum, Cell-free, body fluids and Cell-culture supernatants; 1999. [ Links ]
11. Brown TM, Chang GJ, Croop CB, Robbins KE, Tsai TF. Detection of Yellow fever virus by polymerase chain reaction. Clinical and Diagnostic Virology 1994; 2: 41-51. [ Links ]
12. QIAGEN. The QlAexpressionist. A handbook for high-level expression and purification of 6xHis-tagged proteins. Third ed. 1997. [ Links ]
13. Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol 1982; 157: 105-32. [ Links ]
14. Mandl CW, Guirakhoo F, Holzman H, Heinz FX, Kunz C. Antigenic structure of the flavivirus envelope E protein at the molecular level using tick-encephalitis virus as model. J Virol 1989; 63: 564-71. [ Links ]
15. Fonseca BAL, Khoshnood K, Shope RE, Mason PW. Flavivirus, type-specific antigens produced from fusions of a portion of the E protein gen with Escherichia coli trpE gene. Am J Trop Med Hyg 1991; 44: 500-8. [ Links ]
Correspondencla: Carlos Yábar Varas.
Instituto Nacional de Salud.
Calle Cápac Yupanqui 1400, Lima 11, Perú.
Apartado postal 471.
Telf.: (0511) 4719920 - Fax: (0511) 4710179.
E-mail: cyabar@ins.sid.pe

