










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
versión impresa ISSN 1726-4634
Rev. perú. med. exp. salud publica v.19 n.3 Lima jul./set 2002
TRABAJOS ORIGINALES
Diseño y evaluación de tres oligonucleótidos para la detección de Leishmania por PCR
Omar Cáceres Rey1; Ysabel Montoya Piedra1
1 División de Biología Molecular, Centro Nacional de Salud Pública, Instituto Nacional de Salud. Lima - Perú.
RESUMEN
La leishmaniasis afecta la salud pública en 88 países del mundo y representa un serio obstáculo para su desarrollo socioeconómico. Los métodos para diagnosticar la enfermedad toman tiempo y muchas veces son traumáticos para el paciente. Objetivo: Se aplicó PCR como método alternativo para el diagnóstico rápido de esta enfermedad.
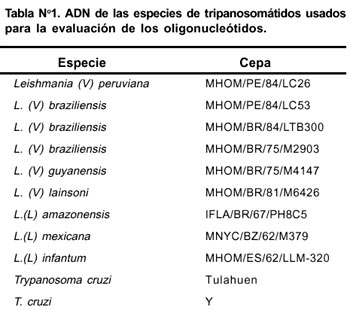
Materiales y métodos: A diferentes especies de Leishmania se les extrajo ADN genómico. Se diseñaron tres oligonucleótidos (LEISH 1, LEISH 2 y LEISH 3) dirigidos al extremo carboxilo terminal de la histona H2B de Leishmania (V.) peruviana cuya secuencia parcial sirvió para diseñar dichos oligos, los cuales fueron probados en un sistema de PCR.
Resultados: Los oligonucleótidos amplificaron exitosamente regiones de 123 pb (LEISH 1 / LEISH 2) y 139 pb (LEISH 1 / LEISH 3) de esta secuencia parcial utilizada. La sensibilidad del PCR fue de hasta 1 fg de ADN purificado de L. (V.) peruviana y de 2 parásitos cuando se realizó la técnica de manera directa. La especificidad fue 100% (solo reconoció a Leishmania y no a Trypanosoma ni a humano). Los oligonucleótidos diseñados también amplificaron todas las especies de Leishmania evaluadas.
Conclusión: El sistema de PCR diseñado puede ser aplicado en la detección del parásito a partir de cualquier tipo de muestra convirtiéndose en un método alternativo de diagnóstico de la enfermedad por su rapidez, especificidad y sensibilidad.
Palabras clave: Leishmaniasis / diagnóstico; Leishmania/genética; Oligonucleótidos / uso diagnóstico; Reacción en cadena de la polimerasa / métodos (fuente: BIREME).
ABSTRACT
Leishmaniasis affects public health in 88 countries of the world and represents an important obstacle for their socioeconomic development. The methods used to diagnose this disease take time and are frequently traumatic to the patient.
Materials and methods Genomic DNA from different Leishmania species were extracted. The carboxi-end terminal of a partial sequence of the H2B histone of Leishmania (V.) peruviana was used to design three oligonucleotides (LEISH 1, LEISH 2, LEISH3) to design a PCR technique as an alternative method to diagnose this disease rapidly.
Results: A PCR was performed with these primers, which amplified succesfully 123bp (LEISH 1/ LEISH 2) and 139bp (LEISH1/ LEISH3) of the partial sequence studied. The sensitivity of direct PCR was appropiate even when 1fg of purified DNA of L. (V.) peruviana and 2 parasites was used. The specificity was 100% in all Leishmania species analyzed and did not recognized neither Trypanosoma nor human DNA.
Conclusions: The oligonucleotides designed could be used in the detection of parasite from different kind of samples like fresh blood with anticoagulant, biopsy and dermal scrapping. PCR has become an alternative method to diagnose leishmaniasis because of its speed, specificity and sensitivity.
Key words: Leishmaniasis/diagnosis; Leishmania/genetics; Oligonucleotides / diagnostic use; Polymerase chain reaction / methods (source: BIREME)
INTRODUCCIÓN
Los protozoos del género Leishmania causan un grupo de enfermedades denominadas Leishmaniasis, cuyo espectro va desde úlceras cutáneas que curan espontáneamente hasta graves afecciones viscerales que pueden comprometer la vida del paciente, existiendo entre estos dos polos una amplia gama de posibilidades clínicas.
La leishmaniasis representa un serio obstáculo para el desarrollo socioeconómico de 88 países, por lo cual la Organización Mundial de la Salud (OMS) ha reconocido a esta enfermedad como un problema de salud pública global1. Son 72 los países en vías de desarrollo afectados por esta enfermedad, entre ellos el Perú. La literatura reporta que existen aproximadamente de 12 a 14 millones de personas infectadas en el mundo y 350 millones de personas en riesgo de contraer la enfermedad, de las cuales 1,5 a 2 millones se infectarán anualmente. De este último grupo, se considera que 500 000 sufrirán leishmaniasis visceral y casi un millón y medio sufrirán leishmaniasis tegumentaria (cutánea).2
En el Perú, esta enfermedad constituye la tercera causa de morbilidad por enfermedades transmisibles después de la malaria y la tuberculosis. En 1999, se reportaron 4 645 casos entre probables y confirmados, en tanto que para el 2002 hasta la semana epidemiológica N° 45 se han reportado 5 998 casos de Leishmaniasis cutánea y 380 casos de Leishmaniasis mucocutánea.3
El diagnóstico de la enfermedad en el laboratorio se realiza usando métodos microscópicos y cultivos in vitro e in vivo a partir de biopsias de la lesión, demostrando la presencia del parásito. Estos métodos son lentos, trabajosos y de limitada sensibilidad, además, requieren personal calificado.4
Numerosos intentos de diagnóstico fueron desarrollados con el fin de evitar procedimientos traumáticos y/o difíciles de realizar; de esta manera surgió el Test de Aglutinación Directa (DAT), el ELISA, el Inmunoblot y la inmunofluorescencia, que permitieron una alternativa rápida y menos costosa herramienta de diagnóstico de la enfermedad; sin embargo, su baja sensibilidad y especificidad, han hecho que estos métodos sean limitados, causando que estas técnicas no sean muy confiables, debido a la presencia de falsos positivos y a las reacciones cruzadas con otras enfermedades como malaria, enfermedad de Chagas, Bartonelosis, entre otras. Además, la limitación se hace evidente cuando la parasitemia en las muestras clínicas es baja, en pacientes inmunosuprimidos o cuando se trata de una Leishmaniasis cuya morfología es atípica.5-7
En el Perú, los procedimientos de diagnóstico incluyen el frotis, el cultivo, la histopatología, la intradermoreacción de Montenegro o leishmanina, el ELISA y la Inmunofluorescencia Indirecta (IFI); todos ellos con elevadas limitaciones a nivel de sensibilidad y especificidad.8
En las últimas dos décadas numerosas técnicas de detección basadas en la tecnología del ADN recombinante han mostrado ventajas sobre las técnicas convencionales. El uso de oligonucleótidos específicos para el ADN del minicírculo en el kinetoplasto (ADNk) por la técnica de la Reacción en Cadena de la Polimerasa (PCR) ha superado a los procedimientos de detección parasitológica convencional convirtiéndose en una herramienta útil para el diagnóstico de esta enfermedad5,9,10. Además, el uso de PCR como herramienta para diagnóstico ha probado ser muy especifico, sensible y rápido10-16 comparado con el cultivo, frotis, IFI, ELISA, DAT e inoculación en animales, pudiendo ser usado en áreas rurales10, además de emplearlo también para tipificación de las especies de Leishmania13.
Basado en estas evidencias, nuestro objetivo fue acondicionar y aplicar la técnica de PCR para diagnosticar la leishmaniasis; para ello se diseñaron oligonucleótidos que al ser usados en la reacción de PCR, amplificaron una región de la Histona H2B del parásito.
MATERIALES Y MÉTODOS
MATERIAL BIOLÓGICO
La cepa de Leishmania empleada para el diseño de los oligonucleótidos fue una cepa de referencia del banco de la OMS codificado como MHOM/PE/84/LC26, esta cepa fue catalogada como Leishmania. (V.) peruviana por estudios con isoenzimas.17, 18 Además, se utilizó ADN extraído de otras cepas de leishmanias y tripanosomas las cuales también fueron referenciales del banco de la OMS (Tabla N° 1). De igual manera se aisló ADN humano proveniente de linfocitos extraídos a partir de sangre perteneciente a un individuo sano.
CRECIMIENTO DE PARÁSITOS
Las cepas de Leishmania fueron cultivadas y mantenidas en medio bifásico19 el cual está constituido por una fase sólida y una fase líquida. La fase sólida está compuesta por 4% w/v de Agar Base Sangre (Difco) suplementada con 15% de sangre desfibrinada de conejo, obtenida por punción cardiaca; la fase líquida está compuesta por NaCl 0,85% estéril, el cual se agrega al medio antes de cultivar a los parásitos. Para evitar el crecimiento bacteriano se agregó a la fase líquida una solución de antibióticos que consta de penicilina sódica y sulfato de estreptomicina, ambos a una concentración de 750 ug/mL. Para controlar la contaminación por hongos o levaduras se utilizó 5-Fluorocitosina al 1% (Sigma). Los parásitos fueron cultivados y mantenidos a 28°C. Cada cuatro o seis días se renovaron los cultivos dependiendo del crecimiento del parásito. Las cepas mantenidas en tubos de ensayo fueron crecidas en frascos de cultivo de 500 mL conteniendo el medio bifásico (30 mL de agar sangre y 5 mL de sobrenadante de cultivo). Los cultivos fueron mantenidos por siete días hasta que los parásitos alcanzaron la fase estacionaria temprana de crecimiento.
OBTENCIÓN DE PARÁSITOS A PARTIR DE CULTIVO
A los parásitos crecidos en los frascos de cultivo, se les agregó 5 mL de solución salina estéril, luego estos frascos se agitaron ligeramente para desprender los parásitos de la fase sólida; la fase líquida fue filtrada por una doble capa de gasa para atrapar el debris del medio de cultivo, sobre tubos de centrifugación fríos. A los tubos se les agregó 5 mL de solución salina y después se centrifugaron a 4°C (IEC Centra MP 4R) a 5000 r.p.m. por 15 min. El sobrenadante fue descartado agregándose al pellet 10 mL de solución salina fría, se volvió a suspender el pellet suavemente y se centrifugó como la vez anterior. Este lavado se repitió por tres veces más, luego del cual nuevamente se suspendió el pellet en 1 mL de solución salina fría, se centrifugó a 10 000 r.p.m. por 1 minuto, se descartó el sobrenadante y se guardó el pellet a -70°C.
Una alícuota (10 µL) de la suspensión final se diluyó hasta 10-3, se tomaron 10 µL de la última dilución y se colocaron sobre una cámara de Neu Bauer para contar los parásitos.
AISLAMIENTO DE ÁCIDOS NUCLEICOS DE PARÁSITOS
El pellet de parásitos (2 x108) fue descongelado y tratado según protocolo descrito20. Brevemente: los parásitos fueron suspendidos una vez más en 10 mL de buffer de digestión (100 mM EDTA, 10 mM Tris-HCl pH 7,5, 100 Mm NaCl), se le añadió SDS al 1% y proteinasa K a una concentración final de 1mg/mL, la solución se incubó a 55 o C por cinco horas en un baño María con agitación suave ocasional. Posteriormente, se añadió ARNasa a una concentración final de 100 µg/mL y se incubó a 50 °C por 2 horas más.
A la solución anterior se le añadió 10 mL de fenol / cloroformo/ alcohol isoamílico (25:24:1) mezclándose por inversión cuidadosamente, se centrifugó a 1 2000 r.p.m. por 10 minutos y a 4°C, se repitió la extracción una vez más y luego solamente con cloroformo. A la fase acuosa de la última extracción se le agregó 0,1 volúmenes de acetato de sodio 3M y 2,5 volúmenes de etanol absoluto helado. El ADN se dejó precipitar a –20°C por 2 horas, luego los tubos se centrifugaron a 1 2000 r.p.m. por 30 min. y a 4°C. El pellet se lavó 2 veces con etanol al 70% y se dejó secar a 40°C hasta que este se evaporó totalmente. Finalmente, el pellet fue nuevamente suspendido en 10 mL de buffer TE (10 mM Tris-HCl pH 8,0, 1mM EDTA) y almacenado a 4°C.
La concentración del ADN extraído fue cuantificada usando un espectrofotómetro UV/Visible (Spectronic 21D), para ello se agregó 20 µL de ADN a 100 µL de NaCl 2N y se completó con agua bidestilada hasta un volumen final de 2mL Sé leyó la absorbancia a 260 y a 280 nm.
Se asumió que para un ADN de doble hebra 1 D.O 260 es igual a 50 µg/mL de ADN. La pureza del ADN extraído se obtuvo de la relación de D.O 260nm/280nm el cual debe ser mayor de 1,7 21 .
DISEÑO DE OLIGONUCLEÓTIDOS
Se eligió una secuencia parcial de 189 nucleótidos que corresponde al extremo carboxilo terminal del gen de la histona H2B de L. (V.) peruviana 22 y que está clonada en el plásmido pUC19. Para el diseño de los oligonucleótidos se usó el software OLIGO versión 3,0, tomándose en consideración las secuencias homólogas de la histona H2B de L. peruviana, L. enriettii, T. Cruzi y la humana 22-25. El software nos permitió calcular y controlar ciertos parámetros de los oligonucleótidos como el tamaño del producto amplificado,ΔG de estabilidad interna de cada oligonucleótido, contenido de guaninas y citosinas, temperatura de hibridación o annealing (Th), temperatura de fusión (Tm), formación de estructuras secundarias, falso apareamiento de cada oligonucleótido (false priming) entre otros. Se diseñaron tres oligonucleótidos: LEISH 1, LEISH 2 y LEISH 3 (Tabla N° 2) con los cuales se formaron dos juegos de amplificación (LEISH 1 / LEISH 2 y LEISH 1 / LEISH 3). Los oligonucleótidos diseñados fueron sintetizados químicamente (Integrated DNA Technologies, Inc. USA).

REACCIÓN EN CADENA DE LA POLIMERASA (PCR)
Las amplificaciones para la optimización de los diversos parámetros necesarios para la estandarización del PCR fueron realizadas según lo descrito.26,27 La concentración final de la reacción de PCR fue de 1X para el buffer, 200 uM para la mezcla de nucleótidos, 0,2 µM para cada uno de los oligonucleótidos, 5 mM de MgCl 2 y 1 U de Taq Gold ADN polimerasa (Applied Biosystems) para 20 µL de volumen final de reacción. El PCR fue realizado en un termociclador PERKIN ELMER 9600. El ciclo de amplificación para ambos juegos de oligonucleótidos fue el siguiente: 1 ciclo de denaturación inicial a 95o C por 10 min, 30 ciclos de 95° C por 35 seg, 61 °C por 45 seg, 72 °C por 30 seg y una extensión final de 72 °C por 10 min. Los viales conteniendo los productos de amplificación fueron guardados a -20 °C.
5 µL de la reacción de PCR fue mezclado con buffer de muestra 6X y colocado en un gel de agarosa al 2% ( BioRad) en buffer TAE 1X junto con un marcador de peso molecular (100 bp ladder, Promega) y sometido a electroforesis horizontal, a 100 voltios constante por 30 min.
El gel fue teñido con una solución de 1 µg/mL de bromuro de etidio por 10 minutos y los productos de amplificación fueron visualizados en un transiluminador de luz UV (UVP) y fotografiados usando una cámara Polaroid R.
Para evaluar la sensibilidad del sistema se hicieron diluciones seriadas de ADN genómico purificado de L. peruviana desde 10 ng a 1 fg y para la evaluación de la especificidad del PCR se utilizó 1 ng de ADN purificado de T. cruzi y de humano.
Para evaluar el sistema con parásitos totales, se utilizaron parásitos obtenidos de cultivo. A partir de 2 x 10 8 individuos se hicieron diluciones seriadas, 10 µL de cada dilución fueron colocados en viales de 0,5 mL y hervidos en baño María por 5 min. Luego de la ebullición, los tubos se enfriaron en hielo por 2 min, luego centrifugados a 10 000 r.p.m. por 2 min, y finalmente se tomó 5 µL del sobrenadante de cada tubo para realizar el PCR descrito anteriormente.
PCR A PARTIR DE SANGRE TOTAL
A un paciente con diagnóstico clínico de leishmaniasis activa y sin tratamiento, con frotis positivo y confirmado por IFI en el Laboratorio de Parasitología del Instituto Nacional de Salud (Lima - Perú), se le extrajo 3 mL de sangre periférica en un tubo con EDTA potásico (Venoject II Merck) pro-cediéndose luego a aislar los leucocitos, los cuales fueron contados en una cámara de Neu Bauer.
A la sangre sin coagular se le añadió 3 mL de solución salina estéril fría y se mezcló cuidadosamente. Esta solución fue añadida lentamente a otro tubo conteniendo 3 mL de solución precipitante de eritrocitos (Histopaque 1077 Sigma) fría, formando 2 fases bien definidas. Se centrifugó a 1 600 r.p.m. por 40 min y a 18 °C. Después de la centrifugación se observaron tres fases definidas: La fase superior, de suero mas anticoagulante, fue retirada cuidadosamente (este suero se puede usar en estudios serológicos); la fase media, es una capa delgada a manera de anillo y que está formada íntegramente por leucocitos, fue retirada cuidadosamente y colocada en tubos de microcentrífuga estériles; y la fase inferior, formada por la solución precipitante y los eritrocitos precipitados, que fue descartada.
Los leucocitos fueron precipitados por centrifugación a 3 000 r.p.m. por 10 min. a temperatura ambiente, las células fueron resuspendidas y lavadas con solución salina estéril fría y precipitadas como la vez anterior, repitiéndose este lavado por dos veces más. Finalmente, el pellet fue resuspendido en 100 µL de solución salina estéril fría y guardada a -70 °C.
Para la extracción del ADN, se usó la metodología de extracción con sales de guanidina.28 50 µL del pellet de leucocitos fueron colocados en un tubo de microcentrífuga al cual se le añadió 800 mL del reactivo DNAzol ( Gibco BRL), se homogeneizó por dos minutos y se centrifugó a 11 000 r.p.m. por 10 min. a temperatura ambiente. El sobrenadante fue trasladado a otro tubo al cual se le añadió 500 µL de alcohol absoluto para precipitar el ADN, se dejó en reposo por 3 minutos y se centrifugó a 3 000 r.p.m. por 5 min. El pellet fue lavado dos veces con alcohol al 95% y centrifugado cada vez a 3 000 r.p.m. por 5 min, el ADN fue secado a 40 o C hasta evaporar el alcohol. Finalmente, se resuspendió en 50 µL de agua libre de nucleasas, se cuantificó y guardó a 4 °C.
Se prepararon las condiciones de reacción para PCR tal como se señaló anteriormente, en este caso 3 µL del ADN (aproximadamente 5 ng) extraído de los leucocitos fue usado en la reacción.
Para evaluar la sensibilidad del PCR en sangre, se hicieron diluciones seriadas a partir de 2x10 4 leucocitos/mL y se procedió a realizar el PCR.
RESULTADOS
LOCALIZACIÓN DE LOS OLIGONUCLEÓTIDOS
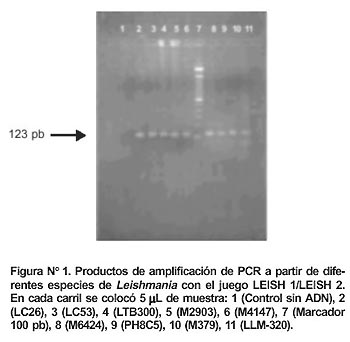
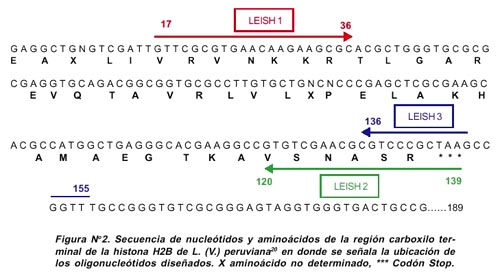
Los oligonucleótidos diseñados exitosamente dieron un producto de amplificación de 123 pb para LEISH 1 / LEISH 2 (Figura N° 1) y de 139 pb para LEISH 1 / LEISH 3. El oligonucleótido LEISH 1 abarcó los nucleótidos del 17 al 36, siendo esta región poco conservada entre L.(V.) peruviana con L.(L.) enrriettii y L. (L) infantum (25% de homología) y algo más conservada entre L. (V.) peruviana con T. cruzi y el humano (30% de homología).
El oligonucleótido LEISH 2 híbrida específicamente entre los nucleótidos 120 y 139, siendo esta región mediana-mente conservada entre L. (V.) peruviana con L. (L.) en-rriettii, L. (L.) infantum y T. cruzi (60%-66,6% de homología), pero divergente entre L. (V.) peruviana y humano (33,3% de homología). El oligonucleótido LEISH 3 hibrida específicamente entre los nucleótidos 136 al 155 y traslapa 5 nucleótidos del oligonucleótido LEISH 2, además, abarcó el codón stop y 8 nucleótidos de la región 3' no traducible (3' UTR) el cual se caracteriza por ser una región muy variable entre las especies. La Figura N° 2 muestra la secuencia parcial de la histona H2B y la localización de los oligonucleótidos diseñados.
SENSIBILIDAD Y ESPECIFICIDAD DEL PCR
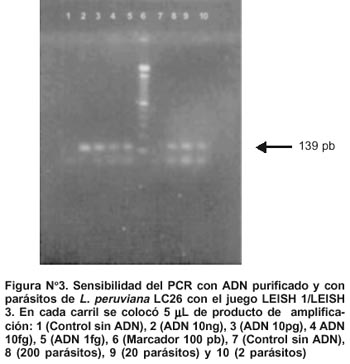
Al probar las diferentes concentraciones de ADN genómico, se logró obtener una amplificación evidente a una concentración tan baja como 1 fg (Figura N° 3). Esto indica que teóricamente el PCR puede detectar hasta un parásito en la muestra.
La sensibilidad del sistema con parásitos totales fue elevada, de modo que se detectó producto de amplificación hasta una concentración tan baja como 2 parásitos presentes en el tubo de reacción (Figura N° 3).
El sistema de PCR amplificó exitosamente todas las cepas de Leishmania evaluadas con ambos juegos de oligonucleótidos (Figura N° 4). No se observó amplificación alguna con Tripanosoma, ni con humano.

SENSIBILIDAD DEL PCR EN SANGRE
El PCR diseñado utilizando el juego LEISH 1/ LEISH 3, produjo una amplificación exitosa hasta una dilución que corresponde a 2,5 leucocitos/mL (Figura N° 5).
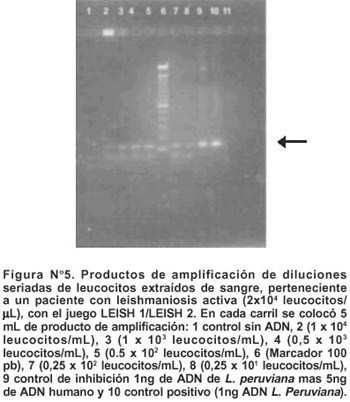
EVALUACIÓN DEL PCR CON MUESTRAS
Muestras de sangre enviadas desde los laboratorios referenciales de Huánuco, Cajamarca y Puno fueron procesadas dentro de las 24 horas de ser recibidas en el laboratorio, realizándose el PCR a un total de 18 muestras, 5 de las cuales fueron positivas por frotis, todas fueron positivas por ELISA al evaluar el suero obtenido durante el proceso de aislamiento de leucocitos, y sólo 4 fueron positivas a PCR.
DISCUSIÓN
Al diseñar los oligonucleótidos se tuvo cuidado para que éstos hibriden específicamente al ADN molde, para lograr esto se tuvo en cuenta la divergencia a nivel de nucleótidos entre la secuencia parcial de L. (V.) peruviana y las secuencias comparadas de L. (L.) enriettii, L. infantum, T. cruzi y humano (Figura N° 6).
La región donde hibridan los oligos LEISH 2 y LEISH 3 es parcialmente conservada entre las Leishmanias y T. Cruzi (60%-66,6% de homología), mientras que con la secuencia de humano es muy divergente (33,3% de homología).
Como se observa, la probabilidad de que los oligos reconozcan las secuencias que no sean de L. (V.) peruviana es muy alta, por lo que la especificidad de estos oligonucleótidos se logra elevar al aumentar la temperatura de hibridación, haciendo así más riguroso su apareamiento con la hebra molde apropiada.
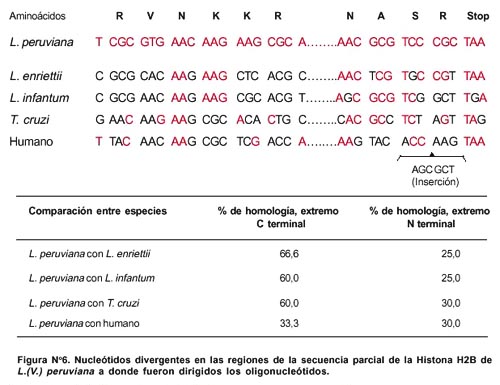
El oligonucleótido LEISH 1 híbrida en una región cercana al extremo amino terminal de la secuencia de la histona (Fig. 1). Esta región es muy divergente entre L. (V.) peruviana con L. Infantum, L. (L.) enriettii, T. cruzi, y humano (25%-30% de homología). Este oligo sería responsable de la elevada especificidad de nuestro PCR (Figura N° 6).
Probablemente, el componente más crítico y el que demanda más tiempo para la optimización de la especificidad de un PCR es la elección de la temperatura de hibridación. Los oligonucleotidos deben realizar una búsqueda genómica hasta que encuentren el sitio complementario en la hebra molde para luego hibridar26.
Entre otras cosas, la probabilidad de una hibridación exitosa en los primeros ciclos del PCR y la especificidad de la hibridación del oligonucleótido depende de la temperatura, del tiempo para la búsqueda genómica, el número de sitios complementarios del oligonucleótido al ADN molde (complementariedad), el diseño del oligonucleótido en sí y el número de copias de la región a amplificar pues, a más copias, más es la probabilidad de una amplificación exitosa29.
Se esperaba que el PCR fuera específico solo para L. (V.) peruviana; sin embargo, al evaluar el PCR con el par LEISH 1/ LEISH 2 se logró una amplificación solo con L. (V.) peruviana y no con T. cruzi ni humano, pero observamos además amplificación exitosa con L. (V.) braziliensis y con las demás especies de Leishmania evaluadas. Probablemente este comportamiento se debe a que los genes de las histonas son altamente conservadas en todas las especies, con algunas divergencias, favoreciendo la hibridación de los oligos 30 .
Cuando se evaluó el juego LEISH 1/ LEISH 3, se observó una amplificación en todos los casos excepto cuando se trató de L. (L.) amazonensis y L. (L.) mexicana donde la amplificación fue más tenue y presentó además una banda de aproximadamente 400 pb (dato no mostrado). Este comportamiento nos indica que estas dos especies presentan una divergencia más marcada en la secuencia de nucleótidos donde se hibridan los oligos, haciendo que el PCR sea poco favorable.
El PCR con el par LEISH 1/ LEISH 3 abriría la posibilidad de que si se ajustan los parámetros del PCR, principalmente la temperatura de hibridación, podría diferenciar a las leishmanias del subgénero Viannia del subgénero Leishmania, a donde pertenecen L. (L.) amazonensis y L. (L.) mexicana. Este resultado podría complementar los resultados obtenidos por otros investigadores, que también pueden discriminar estas especies.10,13,31 La diferencia radica en que estos trabajos fueron realizados amplificando regiones conservadas o variables de los minicírculos del ADNk, que como se sabe existen en gran número y por lo tanto hay muchas copias de las regiones a amplificar existiendo más oportunidad de una amplificación exitosa, comparado con las copias del gen de la histona H2B (20 copias en T. cruzi, 3 copias en tandem en L. (L.) enriettii).
Existen trabajos que hablan de la utilidad del PCR para diagnóstico en sangre circulante tanto para leishmaniasis visceral como leishmaniasis cutánea11,14,32,33 por ello se realizó un ensayo piloto donde evaluamos nuestro PCR con leucocitos aislados de sangre para que pueda ser utilizado para diagnosticar la enfermedad tempranamente y evitar los procedimientos convencionales los cuales son dolorosos y demandan tiempo.5 La dilución de leucocitos hasta donde el PCR logró detectar al parásito, nos indicó que el PCR podría ser utilizado para diagnosticar la enfermedad a partir de muestras sanguíneas. Al evaluar nuestro PCR con muestras de sangre de pacientes con lesiones leishmánicas, se encontró un bajo número de positivos, esto puede ser explicado entre otras cosas: por la demora en ser enviada la sangre al laboratorio desde el momento de la toma de muestra (> 5 días) y al descuido en no ser mantenidas en cadena de frío durante su transporte, ocurriendo la lisis del parásito y por consiguiente, la degradación del ADN.
Se debe considerar además que faltan estudios seriados que determinen si la presencia de macrófagos parasitados en sangre están de forma permanente en el tiempo o están sujetos a fluctuaciones34 por lo que si no hay correspondencia entre el PCR y la historia clínica es recomendable repetir el estudio y aislar el parásito por métodos convencionales5,16.
Finalmente, el sistema de PCR diseñado ha probado ser útil para detectar Leishmania tanto a partir de ADN purificado como directamente de parásitos evitando la necesidad de aislar el ADN, ambos con una elevada sensibilidad y especificidad pudiendo ser utilizado como diagnóstico rápido y confiable. Sin embargo, el PCR no debe ser utilizado como técnica rutinaria pues a pesar de su amplia difusión y aplicación sigue siendo una técnica costosa, pero su utilización esta plenamente justificada para confirmar cuadros clínicos o cuando las técnicas convencionales fallan en la detección del parásito, tal como sucede en casos de leishmaniasis atípica o con baja parasitemia o para estudios epidemiológicos, donde la técnica ha probado ser muy valiosa. Otra utilidad muy importante es el uso de PCR para confirmar la eliminación del parásito después del tratamiento.
Estamos haciendo estudios para evaluar la efectividad de este PCR en la detección del parásito a partir de sangre periférica, en vectores y para estudios de epidemiología molecular.
El presente trabajo es uno de los pocos que ha utilizado ADNg para detectar al parásito pues la gran mayoría de la literatura reporta el uso de ADNk teniendo nuestro PCR un comportamiento similar a los PCR dirigidos a ADNk.
AGRADECIMIENTOS
Los autores agradecen el apoyo técnico de Juana Choque por el cultivo y el mantenimiento de algunas cepas de Leishmania y Trypanosoma utilizadas en este trabajo.
REFERENCIAS
1. World Health Organization. Control of the leishmaniasis. Geneva: World Health Organization Expert Comité; 1990. Technical Report of Series N°793. [ Links ]
2. UNDP/World Bank/WHO. WHO report on global surveillance of epidemic-prone infectious Cap. 10: Leishmaniasis and Leishmania/HIV co-infection. Geneva: WHO; 2000. [ Links ]
3. Oficina General de Epidemiología. Resumen epidemiológico semanal SE 45-2002. Lima: OGE; 2002. [ Links ]
4. Grimaldi G Jr, Tesh RB. Leishmaniasis of the new world: current concepts and implications for future research. Clin Microbiol Rev 1993; 6(3): 230-50. [ Links ]
5. Alvar JP. Las leishmaniasis: de la biología al control. Madrid: Ed. Junta de Castilla y León; 1997. [ Links ]
6. Weiss JB. DNA probes and PCR for diagnosis of parasitic infections. Clin Microbiol Rev 1995; 8(1): 113-30. [ Links ]
7. Piarroux R, Azaeiz R, Lossi AM, Reynier P, Muscatelli F, Gambarelli F, et al. Isolation and characterization of a repetitive DNA sequence from Leishmania infantum: development of a visceral Leishmaniasis polimerase chain reaction. Am J Trop Med Hyg 1993; 49(3): 364-9. [ Links ]
8. Instituto Nacional de Salud. Manual de procedimientos de laboratorio para el diagnóstico de leishmaniasis. Lima: INS; 1995. Serie de Normas Técnicas N° 13. [ Links ]
9. Ashford DA, Bozza M, Freire M, Miranda JC, Sherlock I, Eulalio C, et al. Comparison of the polimerase chain reaction and serology for the detection of canine visceral leishmaniasis. Am J Trop Med Hyg 1995; 53(3): 251-5. [ Links ]
10. López M, Inga R, Cangalaya M, Echevarría J, Llanos-Cuentas A, Orrego C, et al. Diagnosis of Leishmania using the polimerase chain reaction: a simplified procedure for field work. Am J Trop Med Hyg 1993; 49(3): 348-56. [ Links ]
11. Belli A, Rodríguez B, Avilés H, Harris E. Simplified polymerase chain reaction detection of the New World Leishmania in clinical specimenes of cutaneous leishmaniasis. Am J Trop Med Hyg 1998; 58(1): 102-9. [ Links ]
12. Rodríguez N, Guzmán B, Rodas A, Takiff H, Bloom BR, Convit J. Diagnosis of cutaneous leishmaniasis and species discrimination of parasites by PCR and hybridization. J Clin Microbiol 1994; 32(9): 2246-52. [ Links ]
13. Degrave W, Fernándes O, Thiemann O, Wincker P, Britto C, Cardoso A, et al. Detection of Trypanosoma cruzi and Leishmania using the polymerase chain reaction. Mem Inst Oswaldo Cruz 1994; 89(3): 367-8. [ Links ]
14. Piñero J, Martínez E, Pacheco R, Aragón Z, De Armas F, Del Castillo A, et al. PCR-ELISA for diagnosis of muco-cutaneous leishmaniasis. Acta Trop 1999; 73: 21-9. [ Links ]
15. Lugo de Yarbuh A, Premoli-de-Percoco G, Valera M. Localized cutaneous leishmaniasis using polymerase chain reaction: a venezuelan family report. Parasitología al día 1997; 21: 71-5. [ Links ]
16. Piarroux R, Gambarelli F, Dumon H, Fontes M, Dunan S, Mary C, et al. Comparison of PCR with direct examination of bone marrow aspiration, myeloculture and serology for diagnosis of visceral leishmaniasis in immunocompromised patients. J Clin Microbiol 1994; 32(3): 746-9. [ Links ]
17. Dujardin JC, Llanos-Cuentas A, Cáceres A, Arana M, Dujardin JP, Guerrini F, et al. Molecular kariotype variation in Leishmania (Viannia) peruviana: indication of geographical populations in Peru distributed along a north-south cline. Ann Trop Med Parasitol 1993; 87(4): 335-47. [ Links ]
18. Arana M, Evans DA, Zolessi A, Llanos-Cuentas A, Arévalo, J. Biochemical characterization of Leishmania (Viannia) braziliensis y Leishmania (Viannia) peruviana by isoenzyme electrophoresis. Trans R Soc Trop Med Hyg 1990; 84: 10148-52. [ Links ]
19. Walton BC, Shaw JJ, Lainson R. Observations on the in vitro cultivation of L. braziliensis. J Parasitol 1977; 63: 1118-9. [ Links ]
20. Maizels RM, Blaxter ML, Robertson BD, Selkirk ME. Parasite antigens, parasite genes: a laboratory manual for molecular parasitology. Cambridge: Cambridge University Press; 1989. [ Links ]
21. Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. New York: Cold Spring Harbor Laboratory Press; 1989. [ Links ]
22. Montoya Y. Molecular analysis of antigen genes in Peruvian leishmaniasis. (Tesis Doctoral). London: University of Cam-bridge; 1993. [ Links ]
23. Genske JE, Cairns BR, Stack SP, Landfear SM. Structure and regulation of histone H2B mRNAs from Leishmania enriettii. Mol Cell Biol 1991; 11(1): 240-9. [ Links ]
24. García-Salcedo JA, Oliver JL, Stock RP, Gonzáles A. Molecular characterization and transcription of the histone H2B gene from the protozoan parasite Trypanosoma cruzi. Mol Microbiol 1994; 13(6): 1033-43. [ Links ]
25. Zhong R, Roeder RG, Heintz N. The primary structure and expression of four cloned human histone genes. Nucleic Acid Res 1983; 11(21): 7409-25. [ Links ]
26. Rolfs A, Schuller I, Finckh V, Weber-Rolfs I. PCR: clinical diagnostic and research. Berlin: Ed. Springer-Verlag; 1992. [ Links ]
27. Saiki R, Gelfand D, Stoffel S, Scharff S, Higuchi R, Horn G, et al. Primer directed enzymatic amplification of DNA with thermostable DNA polymerase. Science 1988; 239: 487-91. [ Links ]
28. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate - phenol - chloroform extraction. Ann Biochem 1987; 162(1): 156-9. [ Links ]
29. Atlas RM, Bej AK. Polymerase chain reaction, En: Methods for general and molecular bacteriology. Washington D.C.: American Society for Microbiology; 1994. [ Links ]
30. Schumperli D. Cell-cycle regulation of histone gene expression. Cell 1986; 45(4): 471-2. [ Links ]
31. Eresh S, Mc Callum SM, Barker DC. Identification and diagnosis of Leishmania mexicana complex isolates by polymerase chain reaction. Parasitology 1994; 109: 423-33. [ Links ]
32. Ravel S, Cuny G, Reynes J, Veas F. A highly sensitive and rapid procedure for direct PCR detection of Leishmania infantum with human peripheral blood mononuclear cells. Acta Trop 1995; 59: 187-96. [ Links ]
33. Smyth AJ, Ghosh A, Hassan MdQ, Basu D, De Bruijn MHL, Adhya S, et al. Rapid and sensitive detection of Leishmania kinetoplast DNA from spleen and blood samples of Kalaazar patients. Parasitology 1992; 105: 183-92. [ Links ]
34. Bozza M, Fernándes O, Degrave WM, Lópes UG. Characterization of Old World Leishmania species using amplified minicircle variable region as molecular probes. Trans R Soc Trop Med Hyg 1995; 89: 333-4. [ Links ]
Correspondencia: Omar Alberto Cáceres Rey.
División de Biología Molecular,
Instituto Nacional de Salud.
Dirección: Cápac Yupanqui 1400. Lima 11 - Perú.
Telf.: (51-1) 4719920. Fax: (051-1)4710179.
E-mail: ocaceres@ins.gob.pe

