










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
Print version ISSN 1726-4634
Rev. perú. med. exp. salud publica vol.24 no.3 Lima July/set 2007
ARTÍCULO ORIGINAL
Variabilidad del gen de la envoltura del VIH-1 en tres grupos humanos con diferentes conductas sexuales de riesgo para adquirir ITS-VIH
Envelope genetic variability of HIV-1 infecting human groups with differents risk sexual behaviour to adquire HIV-STD
Carlos Yábar V1a ; Javier Salvatierra F2b ; Eberth Quijano G2c
1 Laboratorio de Biotecnología y Biología Molecular, Centro Nacional de Salud Pública, Instituto Nacional de Salud. Lima, Perú.
2 Centro de Referencia para ETS Alberto Barton, Dirección de Salud I Callao. Callao, Perú.
a Biólogo, magister en bioquímica y biología molecular
b Médico, magister en salud reproductiva, género y sexualidad
c Médico infectólogo.
RESUMEN
Objetivo: Determinar la variabilidad genética del gen de la envoltura porción C2-V3-C3 (env) del VIH-1 infectando grupos humanos con diferente conducta sexual de riesgo para adquirir ITS-VIH. Materiales y métodos: Se seleccionaron 50 sujetos infectados con VIH-1 de los cuales 19 fueron hombres trabajadores sexuales (HTS), 8 mujeres trabajadoras sexuales (MTS) y 23 sujetos heterosexuales (SH). Se realizó la extracción de ADN genómico y la amplificación del gen env por PCR. Se identificó el subtipo genético por ensayo de movilidad de heterodúplex (HMA) y se confirmaron los resultados por análisis filogenético. Asimismo, se realizó el análisis de recombinación intragenética, diversidad y distancia genética en las tres poblaciones. Resultados: Se amplificó el gen env en 49 muestras (98%) y se logró secuenciar el fragmento en 40 de ellas. Se observó que el 97,5% de las muestras de VIH fueron subtipo B mientras que una muestra no pudo ser clasificada filogenéticamente. Asimismo, se encontraron pequeños tramos de recombinación en el gen env de VIH en MTS (33%), HTS (43%) y SH (45%). El mayor índice de diversidad de nucleótidos (Pi) de env se encontró entre las muestras de VIH provenientes de SH y HTS (0,12 y 0,13 respectivamente). Conclusiones: Se encontró una mayor variabilidad genética del gen env de VIH-1 en las poblaciones de HTS y SH, sin embargo, el subtipo genético y la frecuencia de recombinación de este fragmento genético fue similar en los tres grupos estudiados.
Palabras clave: VIH; Variabilidad genética; Grupos de riesgo; Genotipificación (fuente: DeCS BIREME).
ABSTRACT
Objectives: To determine the genetic variability of C2-V3-C3 envelope genetic portion (env) of HIV-1 from human groups showing different sexual behavior to acquire STD/HIV. Material and methods: Fifty HIV-infected subjects were selected, which nineteen of them were male sex workers (MSW), eight female sex workers (FSW), and twenty three heterosexual subjects (HS). DNA genomic was extracted and the gen env was amplified by PCR. Genetic subtype as identified by heteroduplex mobility assay (HMA) and confirmed by phylogeny. Likewise, intragenic recombination, diversity and genetic distance were analyzed for the three populations. Results: Forty nine (98%) samples were successfully amplified by PCR but only forty were sequenced. HMA and phylogeny analysis revealed that 97.5% of HIV-1 samples were subtype B, but only one sample remain unclassifiable. Likewise, short recombinant regions in gene env were found from FSW (33%), MSW (43%) and HS (45%). Finally, HIV species infecting MSW and HS showed the highest diversity nucleotide between them (Pi) (0,12 and 0,13 respectively). Conclusion: This study revealed that env gene was highly variable in MSW and HS populations, however genetic subtype and recombination frequency were similar for the three groups.
Key words: HIV; Genetic variability; Risk group; Genotyping (source: DeCS BIREME).
INTRODUCCIÓN
El VIH es uno de los agentes patógenos con mayor tasa de variabilidad genética y con una alta capacidad de adaptación en su hospedero humano 1,2. Esta diversidad se refleja en la existencia de 11 subtipos diferentes de VIH-1 dentro del grupo M1 y hasta 32 especies recombinantes3, de las cuales siete ya han sido han caracterizadas en América 3-5.
La presencia de recombinación es consecuencia de la infección mixta de variantes genéticas del VIH6, evento que ha sido observado principalmente en sujetos con comportamiento sexual de alto riesgo7, 8. De esta manera, el factor humano se convierte en un componente importante en la transmisión, recombinación y expansión de especies virales 3, 9.
Al respecto, existen evidencias que demuestran que algunos grupos humanos con comportamiento de alto riesgo para ser infectados por VIH favorecen la transmisión de algunos subtipos genéticos sobre otros 3, 10-12, produciendo como consecuencia una distribución de variantes genéticas que difiere de una región a otra.
En el Perú, la variante genética de mayor prevalencia es el subtipo B13, aunque otros subtipos como el F, A, C y las formas recombinantes BF y CRF17_BF también han sido reportados en muy baja frecuencia14,15. Es de resaltar que en dichos estudios no se ha observado ninguna correlación entre los grupos que presentaron diferente conducta sexual (población de homosexuales y trabajadoras sexuales) y el subtipo genético. Esto sugiere que para establecer diferencias de variabilidad genética del VIH en estos grupos se requiere profundizar la investigación mediante la detección de eventos de recombinación y diversidad genética.
Por tanto, el objetivo del presente estudio fue determinar la variabilidad genética (subtipos, recombinación y diversidad genética) de la porción C2-V3-C3 del gen de la envoltura (env) de VIH-1 que infecta poblaciones con diferente comportamiento sexual para adquirir VIH / ITS tales como trabajadoras sexuales (MTS), sujetos heterosexuales (SH) y hombres homosexuales que practican el trabajo sexual (HTS). Con relación a los HTS, hemos reportado recientemente que las características del comportamiento sexual de este grupo difieren sustancialmente de las MTS principalmente en lo referente al consumo de estupefacientes y contacto sexual con clientes extranjeros dentro y fuera del país16. Creemos que env de VIH-1 en los HTS podría presentar características genéticas diferentes con relación al VIH de las MTS, lo cual pretende ser demostrado en este estudio.
MATERIALES Y MÉTODOS
DISEÑO
El presente estudio es de tipo retrospectivo y comparativo, el cual fue evaluado y aprobado por los comités de investigación y de ética del Instituto Nacional de Salud. El universo comprendió una población de 153 muestras de sangre congelada, proveniente de los proyectos: Detección de subtipos de VIH-1 en trabajadoras y trabajadores sexuales de Lima y Callao(n = 102) e Identificación molecular de mutaciones puntuales relacionadas a resistencia a drogas en VIH- 1 (n = 51). Ambos proyectos fueron llevados a cabo durante los períodos 2003-2005 y cuentan con ficha de consentimiento informado con la autorización del participante para conservar y analizar sus muestras en estudios posteriores de tipo genético-molecular.
Del universo, se seleccionó un grupo de 50 muestras de sangre, las cuales tenían los datos personales completos (nombre, edad, sexo, lugar de procedencia) de los pacientes, un volumen > 200 μL y eran positivas a PCR para los genes gag, de la proteasa y transcriptasa reversa.
MUESTRAS DE REFERENCIA
Como ADN control del PCR y del ensayo de movilidad de heterodúplex (HMA) se incluyeron plásmidos recombinantes conteniendo la región parcial o completa que codifica las proteínas gp120 y gp160 de VIH-117. Los insertos de cada plásmido correspondieron a cepas de VIH-1 de referencia desde el subtipo A al J. Los plásmidos recombinantes fueron proveídos por el HIV Reagent Program del National Institute of Health (NIH) de los EEUU.
EXTRACCIÓN DE ADN
Para realizar la extracción de ADN se tomó un volumen de 200 mL de sangre periférica y se siguieron los procedimientos recomendados por el fabricante del kit de extracción de ADN a partir de sangre QIABlood (QIAGEN). Los tubos conteniendo la solución de ADN total, fueron almacenados a -20 °C para su posterior uso. Los tubos conteniendo el ADN purificado en solución fueron almacenados a -20 °C para su posterior uso.
PCR PARA LA AMPLIFICACIÓN DEL GEN env
La amplificación del gen env de VIH-1 fue llevado a cabo a través de dos pasos de PCR siguiendo las especificaciones recomendadas por Delwart et al.17 excepto para el caso de las muestras negativas para PCR donde se modificaron las condiciones de algunos parámetros físicos como la temperatura de alineamiento del cebador y tiempo de extensión.
ENSAYO DE MOVILIDAD DE HETERODÚPLEX (HMA)
Los productos de amplificación luego de ser analizados por electroforesis en geles de agarosa fueron seleccionados y sometidos a HMA según las especificaciones de Delwart et al.17. Para tal efecto se prepararon productos de PCR del gen env a partir de cuatro subtipos de referencia A, B, C y F, los cuales fueron mezclados en igual proporción con buffer de alineamiento de heterodúplex 10X. La muestra fue sometida a desnaturalización a 97 °C por tres minutos y renaturalización en un baño de hielo (0 ºC). La identificación de los subtipos se llevó a cabo mediante la visualización de los patrones de banda de ADN que presentaron mayor migración (homodúplex) por electroforesis de acuerdo con las especificaciones recomendadas por Delwart et al.17.
SECUENCIAMIENTO DE ADN
Las reacciones de secuenciamiento de ADN se llevaron a cabo siguiendo el protocolo establecido por los fabricantes del kit de secuenciamiento (Thermo Sequenase Cy5 Dye Terminador Cycle Sequencing Kit). La reacción se llevó a cabo usando entre 3 a 4 mL de producto de PCR (20 – 100 ng de ADN) y empleando el primer ED31 para el gen env.
Cada uno de los tubos fue sometido a las condiciones de PCR para la amplificación del gen env17 exceptuando los pasos de desnaturalización y extensión final. La solución final fue sometida a electroforesis usando el secuenciador automático ALF Express.
Para corroborar la confiabilidad de la lectura de las secuencias de nucleótidos, se repitió hasta tres veces el secuenciamiento de ADN de cada muestra. Por cada repetición se volvió a hacer la extracción de ADN genómico, PCR, Nested PCR, purificación de ADN y el secuenciamiento de ADN propiamente dicho.
DETERMINACIÓN DE RECOMBINACIÓN
Se analizaron secuencias mediante el programa Genotyping tool (http://www.ncbi.nlm.nih.gov/projects/genotyping/formpage.cgi) con el fin de realizar una comparación heurística de la región genética por analizar, con una base de datos de 117 secuencias de referencia de diferentes subtipos. De acuerdo con este programa, es posible calcular la identidad genética de la muestra problema con cada uno de los subtipos de referencia mediante comparación de secuencias de nucleótidos a lo largo de toda la región genética.
De existir alguna región en particular con una alta identidad respecto a un subtipo diferente, el programa mostrará una barra de color diferente. Con el fin de complementar los resultados de este análisis, se realizó la ejecución del programa RDP (Recombination Detection Program versión 1.08)18 el cual a diferencia del Genotyping tool, busca regiones recombinantes por filogenia con cepas de subtipos de referencia y selecciona aquellas candidatas que presenten valores estadísticamente significativos de recombinación. Una ventaja importante de este programa es que permite la combinación y ejecución de hasta cinco métodos diferentes.
ANÁLISIS FILOGENÉTICO
Las secuencias de VIH fueron alineadas usando el programa ClustalW versión 1.8319 cuya información permitió el diseño del árbol filogenético. Para este propósito se usó el método Neighbor-Joining20 aplicando el modelo de Kimura 2 parámetros21 considerando 1000 boostrap como réplicas, omisión de gaps y una distribución gamma de 0,5. Este análisis se realizó usando el programa Mega versión 3.122.
ANÁLISIS DE DIVERSIDAD DE NUCLEÓTIDOS (Pi) Y DISTANCIA GENÉTICA
Con el fin de evaluar cual fue el grado de variabilidad genética del VIH que infectó cada uno de los grupos estudiados se realizó el cálculo de la diversidad de nucleótidos (Pi) del env mediante el uso del programa DAMBE versión 4.5.2723. Para el cálculo de Pi, se realizó una comparación de todas las secuencias y se identificó el número de secuencias diferentes dentro y entre cada población.
Asimismo, se realizó el cálculo de la distancia genética con el fin de identificar el grado de divergencia genética del VIH dentro y entre cada uno de los grupos de riesgo. Para tal propósito se recurrió al programa Mega utilizando el modelo de Kimura 2 parámetros con un boostrap de 1000 réplicas.
ANALISIS ESTADÍSTICO
Se recurrió a la base de datos obtenido de las encuestas realizadas a los participantes con el fin de realizar un análisis de frecuencia de las principales características epidemiológicas de cada población con diferente conducta de riesgo. Entre las principales variables estudiadas se consideró el intervalo de edad, sexo, orientación sexual y contacto sexual con extranjeros. Los datos de frecuencias fueron calculados a través del programa Excel de Windows XP.
RESULTADOS
HTS mostraron mayor frecuencia de contacto sexual con extranjeros que otros grupos
Los datos estadísticos de las muestras seleccionadas en el estudio mostraron que el intervalo de edad comprendido entre los sujetos infectados fue de 18 a 67 años de edad, siendo la población de SH la que presentó un promedio de edad mayor al de los demás grupos de riesgo. De manera importante, el grupo de HTS presentó el mayor de porcentaje de contacto sexual con extranjeros (63%). En este mismo grupo se observó un caso de bisexualidad (5,3%) (Tabla 1).
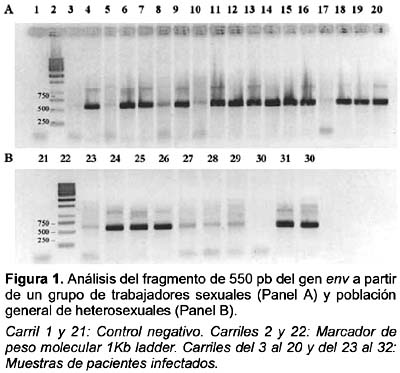
Las modificaciones en las condiciones de amplificación para el gen env de VIH lograron incrementar la sensibilidad de la prueba
De acuerdo con los resultados de Nested – PCR se logró amplificar un producto de aproximadamente 550 pb (ver figura 1). Se consideró la variación de algunos parámetros físicos durante la amplificación como el aumento del tiempo y temperatura de desnaturalización, hibridación y extensión para cinco muestras que salieron negativas usando las condiciones iniciales reportadas por Delwart et al.17, lográndose obtener una sensibilidad de 98% (n = 49) como resultado final. Sólo una muestra, correspondiente a un HTS no se logró amplificar con las modificaciones realizadas en el PCR. Los ensayos de inhibición revelaron que no existió ningún componente inhibitorio (datos no mostrados).
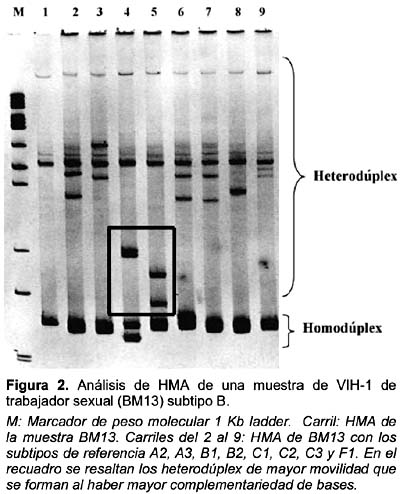
Ensayo de Movilidad de Heterodúplex (HMA) evidenció presencia de subtipo B en la mayoría de las muestras
De acuerdo con los resultados del HMA (figura 2) se observó que 85% (n = 34) de las muestras analizadas fueron subtipo B, mientras que el resto dio resultados indeterminados (Tabla 1). El subtipo de estas últimas muestras fueron luego analizadas por secuenciamiento de ADN (Tabla 2).
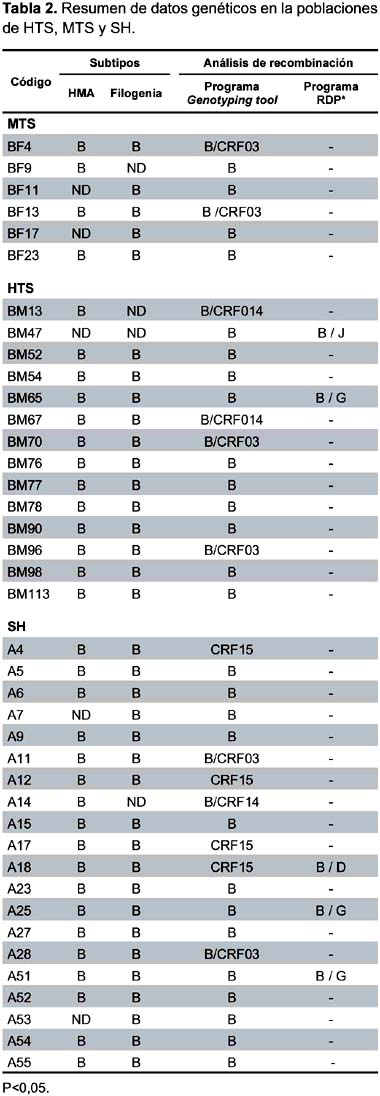
ANÁLISIS DE RECOMBINACIÓN INTRAGENÉTICA
Se intentó identificar evidencias de recombinación en el gen env (recombinación intragenética) usando el programa Genotyping tool. Los resultados revelaron la presencia de tramos de secuencias con regiones altamente idénticas con formas recombinantes de los tipos B / CRF03, B / CRF014 y CRF015 lo cual sugiere la existencia de recombinación intragenética. En total, a través del análisis blast se identificaron dos de seis casos de recombinación en MTS = 33,33%; cuatro de catorce en HTS (28,57%) y seis de veinte en SH = 30,00%, lo cual indica que la presencia de recombinantes entre las tres poblaciones estudiadas es similar (Figura 3 y Tabla 2). Sin embargo, al analizar el tipo de recombinación en cada población, se observó diferencias. Así, B/CRF03 fue visto solamente en MTS, B/CRF014 y B/CRF03 en HTS, mientras que en SH se observó recombinación principalmente con CRF15.
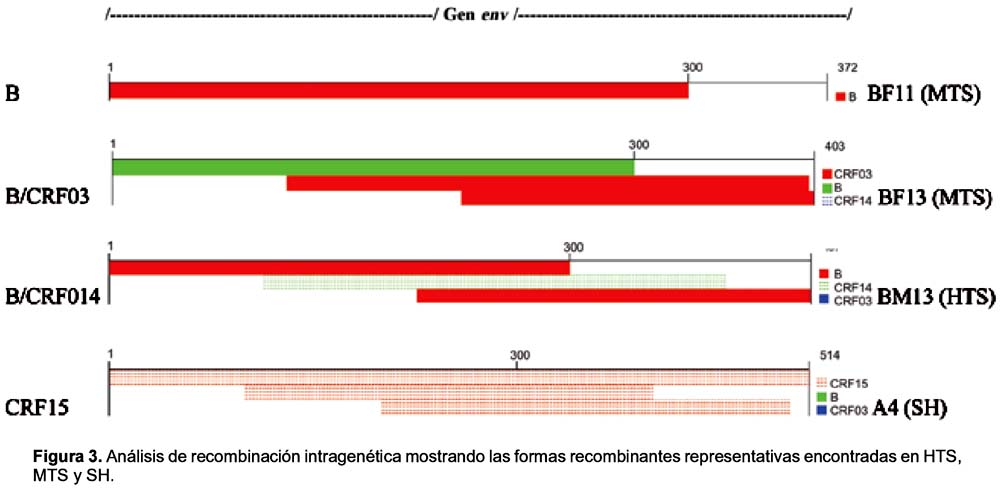
El análisis de recombinación usando el programa RDP versión 1.08 reveló sólo dos casos de recombinación en HTS (B_J, B_G) y tres en SH (B_D, B_G) (p < 0,05)(Tabla 2).
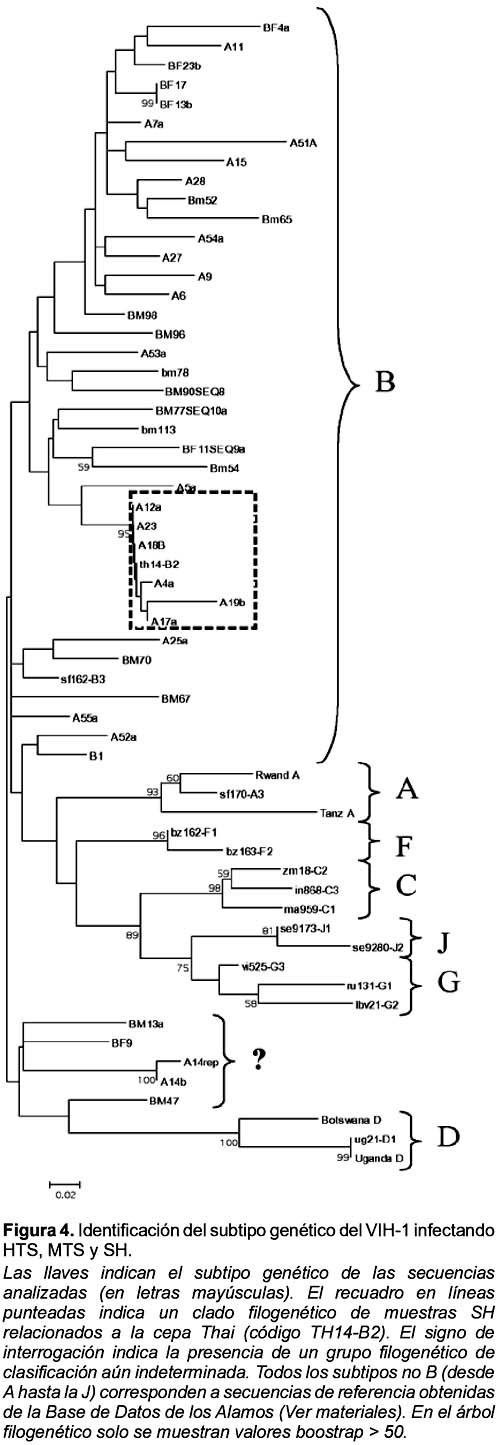
ANÁLISIS FILOGENÉTICO
El análisis filogenético reveló que 90% de las muestras fueron subtipo B (Figura 4) con excepción de un grupo de muestras (BM13, A14, BF9 y BM47) que formaron un grupo filogenético no determinado, el cual se ubicó entre los subtipos B y D. Para corroborar los resultados obtenidos se usó el método de máxima parsimonia, el cual mostró la misma topología confirmando que el subtipo predominante en estas muestras es el B (datos no mostrados). Es de resaltar que 100% de especies de VIH provenientes de HTS y MTS no mostraron una consistente relación filogenética con el subtipo B (valor boostrap < 75, Figura 4). En contraste, un grupo de seis (30%) muestras proveniente de SH formaron un grupo filogenético muy relacionado con la cepa de referencia subtipo B TH14-B2 (valor boostrap = 95%).
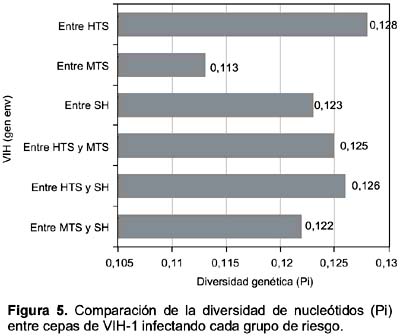
DIVERSIDAD (Pi) Y DISTANCIA GENÉTICA
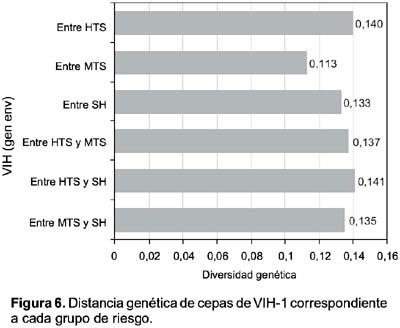
Los datos de análisis de diversidad (Pi) genética del VIH dentro de cada población estudiada revelaron que el mayor índice se encontró entre SH y entre HTS (0,12 y 0,13 respectivamente) mientras que el mínimo valor se encontró entre MTS (0,11) (Figura 5). Es importante señalar que al comparar la diversidad del virus infectando entre cada grupo se observó una mayor diversidad entre HTS y SH (0,13), mientras que el menor valor fue detectado entre MTS y SH (0,12). Con el fin de analizar si la diversidad genética correspondió a la distancia genética del VIH por cada población estudiada se realizó una comparación genética basado en el método de distancias. De acuerdo con los resultados, el gen env de VIH presentó la mayor distancia entre SH y entre MTS (0,13 y 0,14) mientras que entre MTS fue menor (0,11). Al hacer la comparación entre los diferentes grupos se observó que la distancia genética fue similar entre todos ellos (0,14) (Figura 6).
DISCUSIÓN
Nuestros hallazgos sobre la variabilidad genética del gen env (porción C2-V3-C3) de VIH-1 en pacientes peruanos revelan que el subtipo B sigue siendo la variante predominante tal como fue reportado previamente13, sin importar de qué población se trate. Estos hallazgos demuestran que el subtipo B sigue expandiéndose favorablemente en la población peruana y que la existencia de otros subtipos probablemente esté supeditada a cambios en el modo de transmisión viral y en la introducción ocasional de variantes provenientes de otros países. Una evidencia que demuestra la relación existente entre la expansión de la variante genética y el grupo de riesgo fue reportada recientemente entre usuarios de drogas intravenosas (UDI) de Argentina, donde el recombinante BF12 parece circular exclusivamente en este grupo de riesgo en comparación a otras variantes. En el caso de Perú la realidad es totalmente contraria donde la baja o casi nula prevalencia de UDI24 pudiera haber repercutido en la baja frecuencia del subtipo F13-15.
Tomando en consideración que la introducción de nuevos subtipos en el Perú pueda verse favorecida a través del contacto sexual con extranjeros, hemos incluido un nuevo grupo de riesgo formado por HTS16, el cual se trata de una población vulnerable muy poco estudiada en nuestro medio y que se caracteriza por presentar una alta frecuencia de contacto sexual con clientes extranjeros. Nuestros datos de análisis filogenético de env sugieren que el subtipo B de VIH- 1 que circula entre MTS y HTS corresponde a otro linaje respecto a SH. Desde que el contacto sexual con clientes extranjeros ha sido considerado como un factor de riesgo de la transmisión de VIH25, es probable que a través de este medio se haya favorecido la introducción de nuevas cepas de VIH, trayendo como consecuencia la generación de nuevas especies por recombinación.
Este dato se refleja en la presencia de un grupo de muestras clasificadas por env entre los subtipos B y D, que no pudieron ser definidas filogenéticamente. Al respecto, es probable que la presencia de rezagos genéticos del subtipo D en el fragmento de env haya generado tal distribución, en un principio debido a que tanto el subtipo B como el D provienen de un ancestro común26. Sin embargo tampoco se debe descartar la presencia de recombinación e hipermutación (en este caso de tipo B / D) en este grupo, eventos que habrían intervenido en la distribución filogenética de dichas especies virales, tal como se observó en una cohorte de muestras de VIH de Brasil27.
En ese sentido, para que ocurra recombinación B / D es necesaria la introducción del subtipo D en el país por medio del contacto sexual con un portador extranjero. De manera interesante, una de las muestras de código bm47 (Figura 4) fue extraída de un HTS quien reveló haber tenido contacto sexual con diferentes clientes extranjeros aunque sin declarar a qué nacionalidad pertenecieron. Estos datos sugieren que el trabajo sexual con extranjeros dentro y fuera del país podría ser una conducta de riesgo importante para la introducción de nuevos subtipos y formación de nuevas especies de VIH por recombinación.
Con relación a este último punto, el análisis de recombinación intragenética de env reveló la presencia de tramos de recombinación intersubtipo en 17 muestras provenientes de los grupos estudiados. Al respecto es importante mencionar que estos casos en particular no pueden ser considerados como formas recombinantes circulantes de VIH, ya que para ello es necesario caracterizar todo el genoma4,5. Sin embargo, se trata de especies virales que presentan sitios de recombinación en una región genética que codifica parte de la proteína de la envoltura, la cual está implicada en el proceso de entrada del virus en la célula hospedera, así como también en la respuesta inmunológical28. Asimismo, la región analizada comprendió el dominio C2 la cual ha sido caracterizada previamente por presentar la mayor frecuencia de recombinación de VIH, en comparación con otras partes del genoma29. En consecuencia, la presencia de regiones de alta tasa de recombinación en esta proteína es importante ya que puede conferir diferencias en la capacidad de infección, respuesta inmunológica y consecuentemente en la patogenicidad del VIH. Empero, se requieren mayores estudios para corroborar esta hipótesis.
Asimismo, es importante mencionar que los hallazgos de recombinación mostrados en este estudio demuestran indirectamente la existencia de eventos de infecciones mixtas6, lo cual se describe mayormente en sujetos homosexuales con alta promiscuidad sexual y en trabajadoras sexuales7,8. En consecuencia, no debería descartarse la posibilidad de encontrar infecciones mixtas de VIH en la población peruana.
Vale mencionar que se ha demostrado que el gen env de VIH circulando en el grupo de HTS y SH presentó una mayor diversidad y distancia genética que en MTS. Este hecho podría deberse a los múltiples eventos de recombinación observadas en estos grupos, las cuales de acuerdo con otros estudios, favorecen la diversidad del virus26.
Aunque no tenemos evidencias estadísticamente significativas, creemos que algunos factores epidemiológicos observados en HTS, tales como el alto consumo de drogas ilegales, el continuo ejercicio del trabajo sexual en más de 80% de los HTS infectados con VIH y la alta frecuencia de contacto sexual con clientes extranjeros dentro y fuera del país, podrían también favorecer la diversidad del VIH a diferencia de las MTS incluidas en este estudio, donde la conducta sexual no presentó dichas características16.
En conclusión, nuestros datos demuestran que la variabilidad de env de VIH-1 infectando tres grupos humanos con diferente conducta de riesgo es homogénea, principalmente a nivel de subtipos genéticos y recombinación, con excepción de las cepas virales que infectaron HTS y SH donde se encontró una mayor diversidad genética.
Es importante señalar que los datos obtenidos a partir de env no pueden ser extrapolados a la característica genética del todo el virus, puesto que en VIH la presencia de recombinación a lo largo de todo el genoma puede generar una información de variabilidad genética completamente diferente respecto a env. En consecuencia, se recomienda continuar el estudio ampliando el número de marcadores moleculares o bien analizando el genoma completo.
De otro lado, es importante señalar que el número de muestras analizadas en este estudio fue pequeño (n = 50) y por tanto, los hallazgos encontrados no pueden ser considerados como datos representativos para las tres grupos analizados. Por consiguiente, se recomienda incrementar la cantidad de muestras, a fin de conocer con mayor detalle todos los eventos que ocurren durante la evolución del virus en poblaciones con diferente conducta sexual.
AGRADECIMIENTOS
Los autores agradecen la entusiasta participación de los Sres. técnicos de laboratorios Juana Choque Portilla y David García Neyra del Laboratorio de Biotecnología y Biología Molecular del Instituto Nacional de Salud en la realización de las pruebas moleculares.
Agradecemos al NIH AIDS Research and Referent Reagent Program por facilitarnos los cuatro sets de primers para la amplificación de la región p27-p7 de gag y la región C2-V5 del gen env, así como también los subtipos de referencia de VIH-1.
REFERENCIAS BIBLIOGRÁFICAS
1. Requejo HI. Worldwide molecular epidemiology of HIV. Rev Saude Publica. 2006; 40(2): 331-45.
2. Kuiken C, Thakallapalli R, Esklid A, de Ronde A. Genetic analysis reveals epidemiologic patterns in the spread of human immunodeficiency virus. Am J Epidemiol. 2000; 152(9): 814-22.
3. HIV Secuence Database [página de internet]. The Circulating Recombinant Forms (CRFs). New Mexico: Los Alamos National Laboratory; 2006. [Fecha de consulta: noviembre 2006]. Disponible en: http://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html#CRF34
4. Carrion G, Hierholzer J, Montano S, Alava A, Perez J, Guevara A, et al. Circulating recombinant form CRF02_AG in South America. AIDS Res Hum Retroviruses. 2003; 19(4): 329-32. 202-10. Variabilidad genética del VIH
5. De Sa Filho DJ, Sucupira MC, Casiero MM, Sabino EC, Diaz RS, Janini LM. Identification of two HIV type 1 circulating recombinant forms in Brazil. AIDS Res Hum Retroviruses. 2006; 22(1):1-13.
6. Fang G, Weiser B, Kuiken C, Philpott SM, Rowland- Jones S, Plummer F, et al. Recombination following superinfection by HIV-1. AIDS. 2004; 18(2): 153-59.
7. Jost S, Bernard MC, Kaiser L, Yerly S, Hirschel B, Samri A, et al. A patient with HIV-1 superinfection. N Engl J Med. 2002; 347(10): 731-36.
8. Manigart O, Courgnaud V, Sanou O, Valea D, Nagot N, Meda N, et al. HIV-1 superinfections in a cohort of commercial sex workers in Burkina Faso as assessed by an autologous heteroduplex mobility procedure. AIDS. 2004; 18(12): 1645-51.
9. Rambaut A, Robertson DL, Pybus OG, Peeters M, Holmes EC. Human immunodeficiency virus. Phylogeny and the origin of HIV-1. Nature. 2001; 410: 1047-48.
10. van Harmelen J, Wood R, Lambrick M, Rybicki EP, Williamson AL, Williamson C. An association between HIV-1 subtypes and mode of transmission in Cape Town, South Africa. AIDS. 1997; 11(1): 81-87.
11. Avila MM, Pando MA, Carrion G, Peralta LM, Salomon H, Carrillo MG, et al. Two HIV-1 epidemics in Argentina: different genetic subtypes associated with different risk groups. J Acquir Immune Defic Syndr. 2002; 29(4): 422- 26.
12. E spinosa A, Vignoles M, Carrillo MG, Sheppard H, Donovan R, Peralta LM, et al. Intersubtype BF recombinants of HIV-1 in a population of injecting drug users in Argentina. J Acquir Immune Defic Syndr. 2004; 36(1): 630-36.
13. Laguna-Torres VA, Olson J, Montano S, Chauca G, Carrión G, et al. Distribución de los subtipos del VIH-1 en nueve países de América del Sur, 1995-2002. Rev Peru Med Exp Salud Publica. 2005, 22(1): 12-17.
14. Hierholzer J, Montano S, Hoelscher M, Negrete M, Hierholzer M, Avila MM, et al. Molecular epidemiology of HIV type 1 in Ecuador, Peru, Bolivia, Uruguay, and Argentina. AIDS Res Hum Retroviruses. 2002; 18(18): 1339-50.
15. Russell KL, Carcamo C, Watts DM, Sanchez J, Gotuzzo E, Euler A, et al. Emerging genetic diversity of HIV-1 in South America. AIDS. 2000; 14(12): 1785-91.
16. Yábar C, Salvatierra J, Quijano E. Características epidemiológicas del comportamiento sexual en hombres y mujeres trabajadores sexuales infectados con el virus de la inmunodeficiencia humana tipo 1 de Lima y Callao. En: IX Congreso Peruano de Enfermedades Infecciosas y Tropicales - II Congreso Peruano de Control de Infecciones Intrahospitalarias - I Congreso Peruano de SIDA Dr Manuel Huaroto Sedda. [Abstract 84 ] Lima: Sociedad Peruana de Enfermedades Infecciosas y Tropicales; 2005.
17. Delwart EL, Herring B, Rodrigo AG, Mullins JI. Genetic subtyping of human immunodeficiency virus using a heteroduplex mobility assay. PCR Methods Appl. 1995; 4(5): S202-16.
18. Martin DP, Williamson C, Posada D. RDP2: recombination detection and analysis from sequence alignments. Bioinformatics. 2005; 21(2): 260-62.
19. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994; 22(22): 4673-80.
20. Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987; 4(4): 406-25.
21. Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980; 16(2): 111-20.
22. Kumar S, Tamura K, Nei M. MEGA: Molecular Evolutionary Genetics Analysis software for microcomputers. Comput Appl Biosci. 1994; 10(2): 189-91.
23. Xia X, Xie Z. DAMBE: software package for data analysis in molecular biology and evolution. J Hered. 2001; 92(4): 371-73.
24. McCarthy MC, Wignall FS, Sanchez J, Gotuzzo E, Alarcon J, Phillips I, et al. The epidemiology of HIV-1 infection in Peru, 1986-1990. AIDS. 1996; 10(10): 1141- 45.
25. Bautista CT, Sanchez JL, Montano SM, Laguna-Torres VA, Suarez L, Sanchez J, et al. Seroprevalence of and risk factors for HIV-1 infection among female commercial sex workers in South America. Sex Transm Infect. 2006; 82(4): 311-16.
26. Cornelissen M, van den Burg R, Zorgdrager F, Lukashov V, Goudsmit J. pol gene diversity of five human immunodeficiency virus type 1 subtypes: evidence for naturally occurring mutations that contribute to drug resistance, limited recombination patterns, and common ancestry for subtypes B and D. J Virol. 1997; 71(9): 6348-58.
27. Ramos A, Tanuri A, Schechter M, Rayfield MA, Hu DJ, Cabral MC et al. Dual and recombinant infections: an integral part of the HIV-1 epidemic in Brazil. Emerg Infect Dis. 1999; 5(1): 65-74.
28. Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 1998; 280: 1884-88.
29. Galetto R, Moumen A, Giacomoni V, Véron M, Charneau P, Negroni M. The structure of HIV-1 genomic RNA in the gp120 gene determines a recombination hot spot in vivo. J Biol Chem. 2004; 279(35): 36625-32.
Correspondencia:
Carlos Yabar Varas.
Laboratorio de Biotecnología y Biología Molecular, Centro Nacional de Salud Pública, Instituto Nacional de Salud. Lima, Perú
Dirección: Av. Defensores del Morro (Ex Av. Huaylas) 2268, Chorrillos. Apartado Postal Lima 9.
Teléfono: (511) 251-6151 anexos 424 y 546.
Correo electrónico: cyabar@ins.gob.pe , bioyabar@yahoo.es

