










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
versión impresa ISSN 1726-4634
Rev. perú. med. exp. salud publica v.24 n.4 Lima oct./dic. 2007
ARTÍCULO ORIGINAL
Estudio de bioequivalencia del ibuprofeno genérico 400mg tabletas
Bioequivalence study of ibuprofen 400 mg tablets
Ofelia Villalva-Rojas1,a, Miguel Grande-Ortíz1,a, Juan Ortiz1,a, Jacqueline Isasi1,a, Dula Yantas1,a, Víctor Fiestas2,b.
1 Laboratorio de Biodisponibilidad-Bioequivalencia, Centro Nacional de Control de Calidad, Instituto Nacional de Salud. Lima, Perú.
2 Centro Nacional de Salud Pública, Instituto Nacional de Salud. Lima, Perú.
a Químico Farmacéutico, b Médico infectólogo
Estudio financiado por el Instituto Nacional de Salud del Perú.
RESUMEN
Objetivo. Determinar la biodisponibilidad de dos formulaciones de ibuprofeno 400mg tabletas, para establecer si el medicamento multifuente (genérico) es bioequivalente al de referencia (Motrin® 400mg). Materiales y métodos. Se diseñó un estudio abierto, randomizado, cruzado, dos periodos, con siete días de lavado, con 12 voluntarios sanos de ambos sexos, entre 21 y 48 años, quienes ingirieron una tableta del medicamento genérico o de referencia, según randomización, con 200mL de agua. Luego de ingerir el medicamento se colectó 4mL de sangre por voluntario para la cuantificación plasmática de ibuprofeno. Las muestras de plasma se analizaron por cromatografía líquida acoplada al espectrofotómetro de masas (LC-MS/MS) con ionización electrospray ión negativo, aplicando monitoreo de reacción selectiva. La bioequivalencia se determinó con los parámetros farmacocinéticos de área bajo la curva AUC(0-t), AUC(0-∞) y concentración máxima (Cmax). Resultados. Según análisis estadístico, se encontraron: AUCmultifuente(0-t) = 86,85 (μg*h)/ mL, AUCRef.(0-t)= 81,20 (μg*h)/mL, AUCmultifuente(0-∞)= 88,67 (μg*h)/mL, AUCRef.(0-∞)= 82,83(μg*h)/mL, Cmáxmultifuente = 17,70 ug/mL, CmáxRef. =18,09 μg/mL, con rango de 0,93-1,24 para AUC(0-t), 0,93-1,24 para AUC(0-∞) y 0,81-1,19 para Cmax. Conclusión. Los valores encontrados de ibuprofeno están dentro de los requisitos de la OMS y la FDA, para establecer bioequivalencia (0,80–1,25), demostrándose que el ibuprofeno genérico es bioequivalente al de referencia en velocidad y cantidad de ibuprofeno absorbido en el organismo.
Palabras clave: Bioequivalencia; Biodisponibilidad, Farmacocinética; Medicamentos genéricos; Ibuprofeno (fuente: DeCS BIREME).
ABSTRACT
Objective. To determine the bioavailability of two dosage forms of ibuprofen 400mg tablets, for establishing if the multisource (generic) drug is bioequivalent to the reference (Motrin® 400mg tablets). Materials and methods. It was designed an opened study, randomized, two periods, cross over, and seven days washout period, with 12 healthy volunteers (male and female), between 21 and 48 years old, who have taken one tablet of the multisource or reference tablets; according to randomization; with 200 mL of water. After drug intake 4 mL of blood was collected from each volunteer, for quantification of ibuprofen. The plasma samples with ibuprofen and sodium diclofenac (internal standard) were analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS) with negative ion electrospray ionization using selected reaction monitoring. The bioequivalence was established with pharmacokinetics parameters: area under the curve AUC(0-t), AUC(0-∞) and maximum concentration (Cmax). Results. According to statistical analysis were founded: AUCmultisource(0-t) = 86.85 (ug*h)/mL, AUCRef.(0-t) = 81.20 (ug*h)/mL, AUCmultisource(0-∞) = 88.67 (ug*h)/mL, AUCRef.(0-∞) = 82.83 (ug*h)/mL, Cmáxmultisource = 17.70 ug/mL, CmáxRef.=18.09 ug/mL, with intervals of 0.93-1.24 for AUC(0-t), 0.93-1.24 for AUC(0-∞) and 0.81-1.19 for Cmax. Conclusions. The values founded for AUC(0-t), AUC(0-∞) and Cmax are within the established limits by WHO and FDA (0,80 -1,25), so ibuprofen 400mg tables; multisource drug, is bioequivalent to Motrin 400mg tablets, with regard to both the rate and extent of absorption.
Key words: Bioequivalence; Bioavailability, Pharmacokinetics; Drug generic; Ibuprofen (source: DeCS BIREME).
INTRODUCCIÓN
El ibuprofeno es un antiinflamatorio no esteroideo (AINE) derivado del ácido propiónico (ácido débil)1. El tiempo en que alcanza la concentración máxima (Tmáx), tras la administración oral, oscila entre 1-2 h, y la semivida de eliminación es de 2-3 h2. Se excreta rápidamente por la orina, sobre todo en forma de metabolitos y sus conjugados1. Es uno de los antiinflamatorios más consumidos en nuestro país y a nivel mundial, por ser considerado uno de los antiinflamatorios-analgésicosantipiréticos de primera elección, indicado para dolor leve o moderado postoperatorio traumático y en cuadros febriles, tanto en adultos como en niños, además de ser uno de los AINE, con mejor tolerabilidad gastrointestinal y haberse demostrado ampliamente su eficacia y seguridad3.
La incorporación en el mercado farmacéutico de los medicamentos denominados genéricos (multifuentes), presenta un interés económico relevante, ya que el costo de un tratamiento con estos fármacos suele ser inferior al tratamiento con otros productos que contienen el mismo principio activo y en la misma forma farmacéutica, pero con denominaciones comerciales diferentes4. El menor costo se debe a que la inversión económica realizada por el laboratorio farmacéutico para su desarrollo y comercialización del producto multifuente es menor que en el caso de los innovadores (originales)5. Con los multifuentes no es necesario demostrar la eficacia y la relación beneficio/riesgo del producto, solamente demostrar que la curva temporal de niveles plasmáticos del principio activo, contenido en el medicamento genérico es equivalente a la curva temporal obtenida con el medicamento innovador (referencia)4, por lo tanto, para sustituir medicamentos innovadores por multifuentes, se debe contar con evidencia científica, basada en estudios de equivalencia terapéutica in vivo (estudios de bioequivalencia) o in vitro (perfiles de disolución).
El problema actual para todos los involucrados con medicamentos, es saber si dos medicamentos tienen el mismo efecto terapéutico, especialmente si se tiene en cuenta las falsificaciones, el contrabando, las adulteraciones, la gran cantidad de medicamentos multifuentes de dudosa calidad y otros problemas existentes, no sólo en nuestro país, sino a nivel internacional, especialmente en los países tercermundistas, es así, que la implementación de normativas de biodisponibilidad/bioequivalencia en todos los países de la región se hace inaplazable, a fin de garantizar la eficacia y seguridad de todos los medicamentos comercializados6. Por ello, el objetivo del estudio fue determinar la biodisponibilidad de dos formulaciones de ibuprofeno 400mg tabletas para establecer, si el medicamento multifuente (genérico) es bioequivalente al de referencia.
MATERIALES Y MÉTODOS
SUJETOS7-9
Se diseñó un estudio de bioequivalencia (BE) de tipo abierto, randomizado, cruzado, en dos periodos, con siete días de lavado y se ejecutó en octubre de 2006 en una clínica particular de Lima, Perú. Todos los voluntarios aceptaron y firmaron libremente el consentimiento informado, el cual junto al protocolo fueron aprobados por el Comité de Ética en Investigación del Instituto Nacional de Salud6.
Se seleccionó catorce voluntarios sanos de ambos sexos, según los criterios de inclusión y exclusión señalados por la OMS10. Las edades fluctuaron entre 21 y 48 años edad (promedio 31 años), peso entre 52 y 79 kg (promedio 64 kg), talla entre 1,47 y 1,73 m (promedio 1,61 m) y el índice de masa corporal entre 21,41 y 26,70. Todos con resultados normales en los análisis de laboratorio (hematología, bioquímica y análisis de orina) y con resultados negativos para VIH, hepatitis B y C.
PROCEDIMIENTO CLÍNICO10
Los voluntarios permanecieron en ayunas durante diez horas. Cada voluntario ingirió una tableta de ibuprofeno de 400 mg sea el medicamento multifuente o de referencia, con 200 mL de agua sin gas, antes de la toma del medicamento a cada uno se le extrajo 25 mL de sangre para los controles de calidad (QC) y curvas de calibración, luego de la administración del medicamento, se colectó 4 mL de sangre a cada voluntario a los siguientes intervalos: 0,5; 1,0; 1,5; 2,0; 2,5; 3,0; 3,5; 4,0; 5,0, 6,0; 8,0 y 12 horas, utilizando edetato de potasio como anticoagulante. Las muestras de sangre colectadas se centrifugaron a 3500 rpm por diez minutos, a temperatura ambiente y el plasma resultante fue almacenado a -20 °C hasta su análisis1.
Cuatro, seis y diez horas después de ingerir la medicación tomaron sus respectivos alimentos estandarizados. Terminada la última colecta de sangre se dio de alta, previa evaluación médica. Ningún voluntario consumió bebidas alcohólicas, café u otros alimentos que contengan xantinas11.
Toda la etapa clínica estuvo bajo la supervisión de un médico del INS y dos médicos de la clínica, quienes vigilaron la salud de los sujetos durante el estudio.
Cumplido el tiempo de lavado los voluntarios volvieron a ser internados para continuar con el segundo periodo del estudio.
CONTROL DE CALIDAD Y PERFILES DE DISOLUCIÓN
El medicamento multifuente, ibuprofeno 400 mg tabletas, lote 004176, F.V. 04 2009, de origen nacional, fue adquirido en la farmacia de la DIGEMID-MINSA y el de referencia, Motrin® 400 mg tabletas; lote U9553, de laboratorio Pharmacia Upjohn, F.V. 06 2007, droguería Química Suiza. Se usaron estándares primarios USP tanto de ibuprofeno (lote J1E043), como de diclofenaco sódico (lote H0B150), este último como estándar interno (SI)12.
En el Centro Nacional de Control de Calidad del Instituto Nacional de Salud del Perú (CNCC-INS) se realizó el control de calidad para ambos medicamentos, peso promedio (Farmacopea Británica, edición 200513), valoración del principio activo y uniformidad de contenido por variación de peso (Farmacopea de los Estados Unidos, ed 2914) y para la ejecución de los perfiles de disolución y determinación del factor de similitud (f2) se aplicó el Reporte 40 - OMS-200610.
PROCEDIMIENTO ANALÍTICO
Las concentraciones de ibuprofeno se determinaron mediante un método analítico validado, en el equipo de cromatografía líquida acoplado a detector de masas (LCMS/ MS), modelo TSQ Quantum Discovery Max, marca Thermo Finnigan (USA). El ibuprofeno y el diclofenaco se extrajeron del plasma de los voluntarios, tomando 200 μL de plasma y 50 μL de diclofenaco sódico (0,1 ug/mL), más 100 μL de ácido fosfórico (1,4 M), mediante extracción líquida/líquida, con éter dietílico: hexano (80:20 v/v), se evaporó y el residuo se reconstituyó con 200 μL de fase móvil13. Las muestras se procesaron con el software Xcalibur Core Data System – versión 1,4 del equipo LC-MS/MS.
Condiciones cromatográficas. Se usó una columna cromatográfica RP-18 (100mm x 4,6mm i.d.), 3μ, VARIAN, MonoChrom, flujo: 0,30 mL/min, temperatura de la columna 40 °C, temperatura del automuestreador 4 ºC, volumen de inyección 5 μL, y fase móvil, una mezcla de amoniaco:agua:acetonitrilo (0,35mL:200mL:800mL).
Condiciones del espectrofotómetro de masas. Se aplicó ionización electrospray ión negativo, monitoreo de reacción selectiva (SRM); para ibuprofeno SRM 204,91>161,02 y diclofenaco SRM 294,91>249,91.
Validación del método15-17. Se utilizó plasma contaminado con ibuprofeno y SI, se empleó controles de calidad (QC) a tres concentraciones (baja, media y alta); 0,1537; 1,7930 y 17,9300 μg/mL respectivamente, estos QC se utilizaron para determinar los parámetros de validación, a excepción de especificidad en el que se usó plasma normal, hiperlipémico y hemolizado. Para la precisión inter e intradía se usaron QC por quintuplicado. En la curva de calibración se prepararon nueve concentraciones entre 0,0512 μg/μL y 20,4915 μg/μL incluyéndose un estándar blanco y un estándar cero (plasma más SI).
La recuperación de ibuprofeno y diclofenaco fue determinada comparando los valores de los QC frente a las mismas concentraciones en fase móvil.
Para la prueba de arrastre (carry over) se inyectó la concentración más alta de la curva de calibración (20,4915 μg/μL), cuyo cromatograma se comparó con el de plasma blanco.
Las pruebas de estabilidad de ibuprofeno y diclofenaco fueron determinadas tanto para el almacenamiento como para el proceso analítico:
-
La prueba de congelamiento-descongelamiento se realizó con los QC en tres ciclos por 72 horas, cuyas muestras se congelaron a -20 °C y se descongelaron a temperatura ambiente.
-
La estabilidad a corto plazo se realizó a temperatura ambiente durante seis horas, también con los mismos QC.
-
La estabilidad a largo plazo se determinó con los QC almacenados durante sesenta días a -20°C, cuyos resultados se compararon con QC recién preparadas.
-
La estabilidad de las soluciones stock de ibuprofeno (10 μg/mL) y diclofenaco (10 μg/mL), almacenadas durante siete días a temperatura 4 °C, se compararon con muestras frescas a las mismas concentraciones.
-
Así mismo, se determinó la estabilidad de los controles de calidad en el automuestreador a 4 °C por 24 horas.
ANÁLISIS FARMACOCINÉTICO Y ESTADÍSTICO
Fue realizado de acuerdo al método compartimental de Wagner-Nelson. El área bajo la curva (AUC0-t) de la concentración respecto al tiempo hasta el último punto con concentración cuantificable (Ct) se calculó mediante el método de los trapecios. La constante de eliminación (ke) se calculó mediante regresión logarítmica lineal de las concentraciones en la fase de eliminación terminal. El cálculo del AUC hasta el infinito (AUC0-∞) se realizó mediante la adición de la razón Ct/ ke al AUC0-t.
La concentración máxima (Cmáx) y el Tmáx se derivaron de los datos experimentales y el tiempo de vida media se derivó del cálculo ln(2)/ ke1.
Previamente a la aplicación de los cálculos para determinar bioequivalencia, se aplicó el análisis de varianza (ANOVA) a los datos transformados a logaritmo natural, cuantificándose la variabilidad de los factores de secuencia, período y tratamiento en el diseño del estudio.
Se efectuó la evaluación estadística de los datos farmacocinéticos, del AUC0-12, extrapolando el AUC0-∞ y Cmáx con un intervalo de confianza (IC) de 90% para la diferencia entre los valores promedios obtenidos para el medicamento multifuente y el de referencia, usando el programa Microsoft Excel 2003. Estos resultados fueron verificados con el programa STATA 7.0 y el Bioequivalence program for two – period cross – over studies, versión 3.7.
RESULTADOS
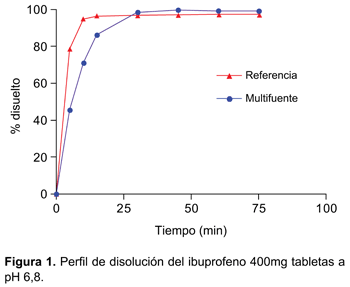
Ambas formulaciones cumplieron con las especificaciones de las obras oficiales en el control de calidad y en los perfiles de disolución se obtuvieron: f2=74,54, en el medio a pH 1,2; f2=58,30 en el medio a pH 4,5 y en el medio a pH 6,8 se observó que a los 15 minutos ambos medicamentos liberaron más de 85% del principio activo (Figura 1), por lo cual no fue necesario aplicar el factor de similitud (f2).
Todos los parámetros de la validación15,18 se encontraron dentro de las especificaciones requeridas, en especificidad los cromatogramas de los tres tipos de plasma no mostraron interferencias. El límite de detección para ibuprofeno fue determinado en 0,0512 μg/mL y el límite de cuantificación en 0,1025 μg/mL, con precisión de 101,3% y exactitud de 107,0%. La precisión intradía mostró un coeficiente de variación de 3,6%, 5,9% y 13,4% para los QC bajo, medio y alto respectivamente, mientras que la precisión interdía presentó un coeficiente de variación de 9,1%, 4,8% y 10,5% para las mismas concentraciones. La exactitud intradía encontrada fue de 96,8%, 98,8% y 105,9%, con una variación de -3,2%; -1,2% y 5,9% para los tres QC. El coeficiente de correlación de la curva de calibración fue de 0,9987.
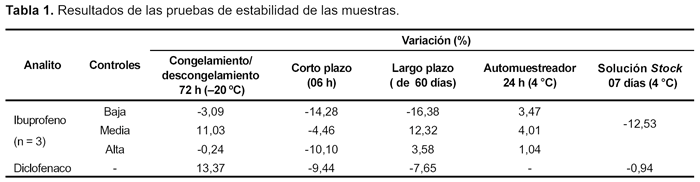
El porcentaje de recuperación para ibuprofeno fue de 86,1%; 102,5% y 115,9% para los tres QC. Los cromatogramas de la prueba de arrastre (carry over) muestran que no existió contaminación del autoinyector. Los resultados obtenidos (Tabla 1) en las pruebas de estabilidad del analito y del SI cumplen las especificaciones (no mayor a 20% con relación a las muestras frescas).
RESULTADOS FARMACOCINÉTICOS
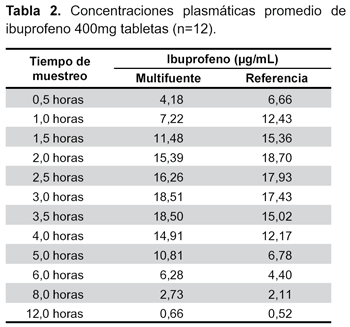
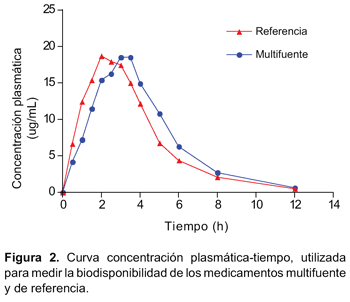
El estudio se llevó a cabo con catorce voluntarios, sin embargo, la evaluación se realizó sólo en doce, debido a que dos de ellos no cumplieron lo establecido por el modelo de Wagner-Nelson. Las concentraciones plasmáticas y los tiempos de muestreo de ambas formulaciones se muestran en la Tabla 2 y la curva de concentración plasmática frente al tiempo en la Figura 2. Se calculó los principales parámetros farmacocinéticos por cada sujeto (Tabla 3).
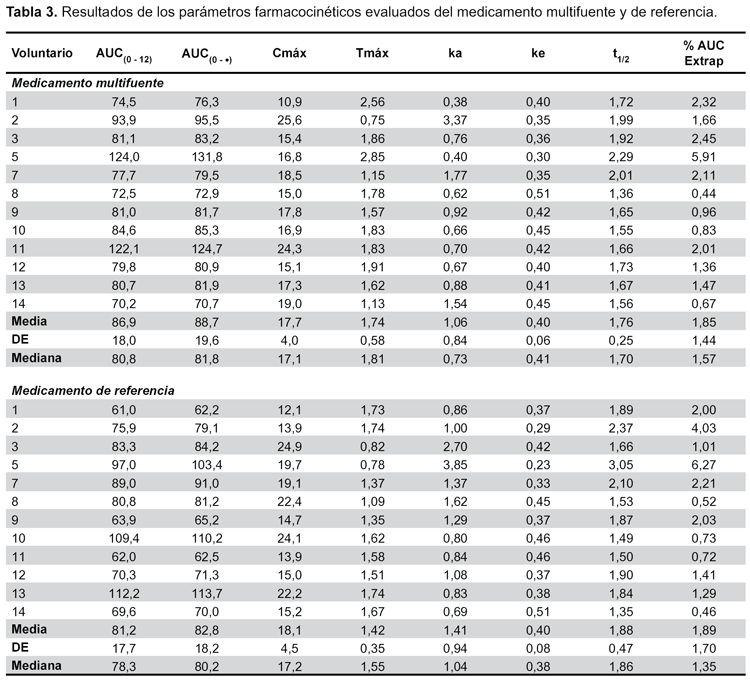
Los resultados del análisis estadístico (ANOVA) mostraron que el punto de estimación, los límites de bioequivalencia de los parámetros farmacocinéticos no fueron afectados por factores de variabilidad secuencia, periodo y tratamiento en el diseño del estudio, de acuerdo con la randomización aplicada.
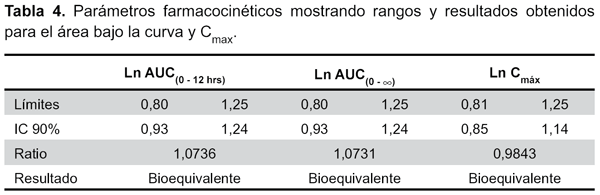
La comparación de las AUC resultó bioequivalente, tanto para el AUC0-12, como para el AUC0-∞, así mismo, comparando Cmáx, lo que muestra la bioequivalencia entre el producto multifuente y el de referencia, cuyos rangos de variabilidad se encuentra dentro de los intervalos de 0,80 a 1,25 para bioequivalencia según la OMS10 y la FDA11 (Tabla 4).
DISCUSIÓN
En muchos países antes de comercializar los medicamentos se exige garantizar la calidad, seguridad y eficacia. Los medicamentos genéricos (multifuente) demuestran la seguridad y eficacia a través de estudios de bioequivalencia con el medicamento innovador, con el fin de garantizar su intercambiabilidad19. En el Perú, actualmente no se exige evidenciar la equivalencia terapéutica para los medicamentos que pretendan la obtención del registro sanitario20, realizándose solamente la vigilancia sanitaria a través de control de calidad post registro.
El presente estudio es de gran importancia para nuestro país, por ser uno de los primeros en su tipo en realizarse para establecer la intercambiabilidad de los medicamentos según las exigencias de la OMS.
En los resultados obtenidos del perfil de disolución según exigencias de OMS se evidenció tanto para la formulación de referencia como para el multifuente que el porcentaje liberado en el medio de disolución pH 6,8 fue superior al 85% a los 30 minutos, cumpliendo con las exigencias de clase I de la clasificación biofarmacéutica (BCS) (disolución rápida). Además el hecho que a los 15 minutos el porcentaje liberado sea superior a 85% no fue necesario realizar cálculo matemático alguno mediante aplicación del modelo independiente factor de similitud f2 (disolución muy rápida según BCS). Tras la administración de las formulaciones de ibuprofeno 400 mg tabletas se obtuvieron concentraciones plasmáticas elevadas en forma rápida, lo que se consiguió gracias a su rápida disgregación y absorción, evidenciándose valores de Tmáx a las tres horas, con vida media de: 1,76h-1, para el multifuente y un Tmáx a las dos horas, con vida media: 1,88h h-1 para el de referencia.
El método LC-MS-MS descrito y usado para la cuantificación provee una alta sensibilidad, especificidad y alto rendimiento en el procesamiento de las muestras, requerido para estudios farmacocinéticos21. Los niveles plasmáticos de ibuprofeno han sido detectados por otros métodos tales como cromatografía líquida de alta presión (HPLC), acoplado a un detector UV. El límite de cuantificación encontrado en el presente estudio fue de 0,10 μg/mL (curva de calibración entre 0,05 – 20,49 μg/mL), menor a otros métodos de HPLC18 (LOQ, 1,56 μg/mL (curva de calibración entre 0,78 – 100 μg/mL). Así mismo, los tiempos de elución para ibuprofeno 1,5 min y diclofenaco sódico 1,6 min fueron menores en el método de LC-MS-MS en relación al método de HPLC; 1,7 min para naproxeno y 3,1 min para ibuprofeno, con un tiempo de corrida analítica; 1,7 veces mayor al método utilizado en este estudio18.
En las condiciones experimentales ensayadas, la disociación inducida por colisión (CID) de ibuprofeno (m/z 205,04) mostrando un fragmento 161,02 y el CID de diclofenaco sódico (m/z 294,0), mostrando un fragmento 249,91 es similar a lo descrito por otros autores12.
Los promedios de porcentaje de AUC extrapolados tanto para el medicamento multifuente (1,85%) y el de referencia (1,89%) se encontraron dentro de los niveles de aceptabilidad descritos por la OMS, no mayor a 20%, lo cual fue corroborado con valores muy similares de AUC0-t y AUC0-∞. Multifuente 88,85 ug/mL*h y 88,67 μg/mL*h respectivamente y para la referencia 81,20 μg/ mL*h y 82,83 μg/mL*h.
En relación con los resultados, no se observó alteraciones analíticas en las constantes usadas debido a la administración del medicamento en estudio, así mismo, no se registró eventos adversos relacionados con ibuprofeno1.
Se evidenció un perfil de concentración plasmática– tiempo semejante y que la razón entre el AUC de concentración de Ibuprofeno en el tiempo y la razón entre la Cmáx obtenida con cada formulación, estuvieron dentro de los límites de aceptación, tanto para los datos experimentales como para los resultantes de su transformación logarítmica.
En conclusión, los resultados del estudio no mostraron diferencias significativas en los niveles de concentraciones plasmáticas después de la administración de las dos formulaciones de ibuprofeno 400 mg tabletas para la velocidad y la cantidad de ibuprofeno alcanzadas en el organismo. Por lo tanto, el diseño del estudio y la aplicación de los protocolos escogidos permitieron demostrar la bioequivalencia entre los productos.
REFERENCIAS BIBLIOGRÁFICAS
1. Farré M, Roset PN, Pascual JA, Abanades S, Menoyo E, Álvarez Y, et al. Estudio de biodisponibilidad en magnitud y velocidad de comprimidos de ibuprofeno. Reumatol Clin. 2005; 1(3): 155-60.
2. Goodman G.A, Goodman LS, Gilman A. Las bases farmacológicas de la terapéutica. 10ª ed. Mexico DF: Editorial McGraw-Hill; 2003. p. 554-57.
3. Diez Domingo J, Planelles Cantarino MV, Moreno Madrid F, Uberos Fernández J, Moreno Martín J, Molina Carballo A, et al. Evaluación de la eficacia antipirética y seguridad de dos formulaciones pediátricas de ibuprofeno. An Esp Pediatr. 2000; 53(5): 436-40.
4. Zapater P, Horga JF. Bioequivalencia y genéricos. Los estudios de bioequivalencia. I. Una aproximación a sus bases teóricas, diseño y realización. Rev Neurol. 1999; 29(12): 1235-46.
5. Magazzini L, Pammolli F, Riccaboni M. Dynamic competition in pharmaceuticals. Patent expiry, generis penetration, and industry structure. Eur J Health Econ. 2004; 5(2): 175-82.
6. Moreno L. Aspectos éticos de los estudios de biodisponibilidad y bioequivalencia de productos farmacéuticos contenidos en las legislaciones de América Latina. Acta Bioeth. 2004; 10(2): 247-59.
7. Villalva O. PRT-CNCC-004: Organización y planificación de estudios de bioequivalencia. Lima: Centro Nacional de Control de Calidad, Instituto Nacional de Salud; 2006.
8. Storpirtis S, Consiglieri VO. Biodisponibilidade e bioequivalência de medicamentos: aspectos fundamentais para o planejamento e execuçaõ de estudos. Rev Farm Bioquim Univ Sao Paulo. 1995; 31(2): 63-70.
9. E uropean Agency for the Evaluation of Medicinal Products, Committee for Propietary Medical Products (EMEA - CPMP). Note for guidance on the investigation of bioavailability and bioequivalence. London: EMEA; 2001. CPMP/EWP/QWP/1401/98 Disponible en: http://www.emea.europa.eu/pdfs/human/ewp/140198en.pdf
10. World Health Organization. WHO Expert Committee on Specifications for Pharmaceutical Preparations. Fortieth Report. Geneva: WHO; 2006. WHO Technical Report Series 937.
11. Food and Drug Administration, Center for Drug Evaluation and Research (FDA-CDER). Bioavailability and bioequivalence studies for orally administered drug products – general considerations. Rockville, MD: CDERFDA; 2002. Disponible en: http://www.fda.gov/cder/guidance/4964dft.pdf
12. Astigarraga RE. Determination of ibuprofen in human plasma by LC-MS-MS using diclofenac as the internal standard. Sao Paulo: Cartesius Analitical Unit, Institute of Biomedical Sciences, Universidade de São Paulo; 2002.
13. British Pharmacopoeia Commission. British Pharmacopoeia. London: The Stationery Office; 2005.
14. US Pharmacopoeia, 29th ed. Maryland: The United States Pharmacopeial Convention; 2006.
15. O rtiz J, Grande M, Isasi J. PRT-CNCC-005: Validación de métodos analíticos para estudios en matrices biológicas. Lima: Centro Nacional de Control de Calidad, Instituto Nacional de Salud; 2005.
16. Food and Drug Administration, Center for Drug Evaluation and Research (FDA-CDER). Bioanalytical method validation. Rockville, MD: CDER-FDA; 2002. Disponible en: http://www.fda.gov/CDER/GUIDANCE/4252fnl.pdf
17. Agência Nacional de Vigilância Sanitária (ANVISA). Resolução RE n° 899: Guía para validação de métodos analíticos e bioanalíticos. Brasilia: ANVISA; 2003.
18. Farrar H, Letzig L, Gill M. Validation of a liquid chromatographic method for the determination of ibuprofen in human plasma. J Chromatogr B. 2002; 780(2): 341-48.
19. Laguna-Goya N, Blázquez-Pérez A, Pozo-Hernández C. Legislación sobre autorización de genéricos. Farm Hosp. 2006; 30(6): 379-84.
20. Perú, Ministerio de Salud. Decreto Supremo 010-97-SA. Reglamento para el Registro, Control y Vigilancia Sanitaria de Productos Farmacéuticos y Afines. Lima: MINSA; 1997.
21. O liveira CH, Abib E, Vannuchi YB, Sucupira M, Ilhia J, De Nucci G. Comparative bioavailability for 4 amoxicillin formulations in healthy human volunteers after a single dose administration. Int J Clin Pharmacol Ther. 2001; 39(4): 167-72.
Correspondencia: Q.F. Ofelia Villalva Rojas, Centro Nacional de Control de Calidad, Instituto Nacional de Salud. Lima, Perú.
Dirección: Av. Defensores del Morro 2268, Chorrillos, Lima.
Teléfono: (511) 467-6696
Correo electrónico: ovillalvar@yahoo.es; ovillalvar@ins.gob.pe

