










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
versión impresa ISSN 1726-4634
Rev. perú. med. exp. salud publica v.26 n.4 Lima oct./dic. 2009
SIMPOSIO: POLÍTICA DE MEDICAMENTOS
Ley N.º 29459 Ley de los productos farmacéuticos, dispositivos médicos y productos sanitarios
Law N° 29459 of pharmaceutical products, medical devices and sanitary products
Víctor Dongo1
1 Médico, Director General de la Dirección General de Medicamentos, Insumos y Drogas, Ministerio de Salud. Lima, Perú.
RESUMEN
La Ley de los Productos Farmacéuticos, Dispositivos Médicos y Productos Sanitarios N.° 29459, publicada en noviembre de 2009, orienta la regulación de estos productos sustituyendo al Capítulo III de la Ley General de Salud N.º 26842. A través de esta Ley se ha modificado los aspectos más cuestionados en la Ley N.° 26842, al establecer requisitos para solicitar el registro sanitario de productos farmacéuticos que son necesarios para garantizar su eficacia, seguridad y calidad, incluyendo también los plazos necesarios para su evaluación y que el costo de la tasa por registro sanitario sea en función de lo que implique otorgar dicho registro, lo cual incluye también las actividades de control y vigilancia sanitaria. Así mismo, restablece la exigencia de la autorización sanitaria de funcionamiento, previa al inicio de las actividades, previa inspección para verificar el cumplimiento de los dispositivos legales vigentes. La Ley incorpora también tres capítulos específicos sobre acceso, uso racional de productos farmacéuticos, dispositivos médicos y productos sanitarios, así como un capítulo de investigación.
Palabras clave: Legislación de medicamentos; Política nacional de medicamentos; Control de medicamentos y narcóticos; Evaluación de medicamentos; Medicamentos esenciales; Comercialización de medicamentos; Perú (fuente: DeCS BIREME).
RESUMEN
The Law of Pharmaceutical Products, Medical devices and Sanitary products N°. 29459, published in November, 2009, guides the regulations of these products, substituting Chapter III of the General Law of Health N°. 26842. Through this law, the most questioned aspects of the Law N°. 26842 have been modified, establishing requisites to apply for the sanitary registry of pharmaceutical products that are necessary to guarantee their efficacy, security and quality, also including the required terms for their evaluation and the cost of the health registration fee must be according to all what implies to give the registry, including control activities and health surveillance. It also resets the requirement of the operating health approval, prior to the initiation of the activities, and having had a previous inspection to verify the accomplishment of the actual legal devices. The law also incorporates three specific chapters about access, rational use of pharmaceutical products, medical devices and health products, as well as a chapter on research.
Key words: Legislation, drug; National drug policy; Drug and narcotic control; Drug evaluation; Drug, essential; Pharmaceutical trade; Peru (source: MeSH NLM).
ANTECEDENTES DE LA REGULACIÓN DEL REGISTRO SANITARIO
Al inicio de la década de 1990, el mercado peruano de medicamentos sufrió cambios radicales; de ser un mercado altamente regulado, con control de precios y múltiples barreras al ingreso (se registraron menos de 200 medicamentos al año) se convirtió en otro sin regulaciones, con precio libre, más aun cuando se promulgó la Ley General de Salud N.° 26842 (1) , cuyo capítulo III, específicamente, en su artículo 50.°, establecía que la inscripción en el Registro Sanitario (RS) de medicamentos era automática, esta se otorgaba con la simple presentación de una declaración jurada sobre la calidad, seguridad y eficacia del producto, adjuntando, entre otros documentos, un certificado de libre venta emitido por la autoridad sanitaria de cualquier lugar del mundo, cuando el producto es importado, es decir, era un registro por referencia, pudiendo ya ser registrado en el país sin la posibilidad de una evaluación de la eficacia y seguridad de los productos ni de las condiciones necesarias para evaluar su calidad.
Asimismo, la Ley estableció que el RS se otorgue en un plazo de siete días; de igual manera, establecía que el costo por concepto de RS no debía exceder del 10% de la UIT (S/. 355,00 nuevos soles) monto que estaba por debajo del promedio internacional y debía servir, además, para cubrir los gastos de control de calidad, significando esto que los costos de análisis de control de calidad eran asumidos por el Estado.
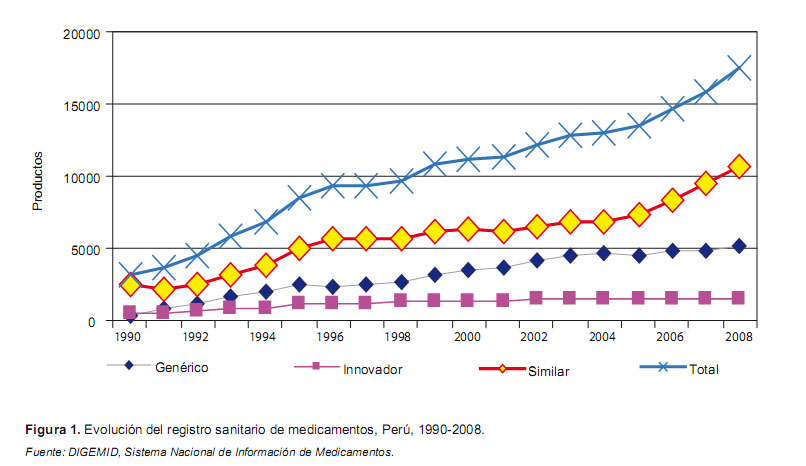
Este hecho explica la gran cantidad de productos registrados durante el período 1990-2008. En efecto, se registró 17 442 medicamentos, de ellos 8,8% (1535) son productos innovadores; la mayoría, 61,4% (10 722) son productos similares de marca y 29,7% (5185) son productos genéricos (Figura 1). De los medicamentos registrados, 48,6% (8482) son de procedencia extranjera y 51,4% (8960) son de origen nacional.
De la misma manera, con estos costos y con los requisitos tan laxos, existía un número elevado de expedientes de RS, que hacía imposible cumplir con el simple trámite administrativo en el plazo establecido, mucho menos cumplir con una evaluación técnica adecuada del producto.
La Dirección de Medicamentos, Insumos y Drogas (DIGEMID) del Ministerio de Salud (MINSA), ante esta falta de regulación y desactualización de las normas, elaboró una propuesta de modifcatoria del Capítulo III de la Ley General de Salud que fue presentada por el MINSA al Poder Ejecutivo y este, a su vez, al Congreso de la República en mayo de 2006. Esta propuesta fue acogida, en gran medida, por el Legislativo, que aprobó la autógrafa que modifcaba la Ley N.º 26842 en su Capítulo III. Sin embargo, el Presidente de la República, observó la mencionada autógrafa y es regresada al Congreso (27 de julio 2006).
La DIGEMID elaboró un nuevo proyecto de modifcatoria del Capítulo III, la Comisión de Salud, Población, Familia y Personas con Discapacidad del Congreso de la República, aprobó el Dictamen 14-2007-2008CSPFPD-CR que modifica la Ley General de Salud, el cual fue consensuado con los aportes de la Presidencia del Consejo de Ministros, Ministerio de Economía y Finanzas, Ministerio de Salud, DIGEMID, Ministerio del Interior, Ministerio de Defensa, Ministerio de la Producción, ESSALUD, Centro Nacional de Control de Calidad del Instituto Nacional de Salud, Instituto Nacional de Defensa de la Competencia y de la Protección de la Propiedad Intelectual (INDECOPI), Asociación de Industrias Farmacéuticas Nacionales (ADIFAN), Asociación Nacional de Laboratorios Farmacéuticos (ALAFARPE), Asociación de Laboratorios Farmacéuticos Latinoamericanos (ALAFAL), Federación Nacional de Químicos Farmacéuticos, Sociedad Nacional de Industrias, Colegio Químico Farmacéutico de Lima y Colegio Químico Farmacéutico Nacional, Colegio Médico del Perú y Cámara de Comercio de Lima y, con fecha 18 de junio de 2008, le dio trámite para su debate y aprobación en el pleno del Congreso.
Con fecha 29 de octubre de 2009 el pleno del Congreso de la República debatió y aprobó el Proyecto de Ley de los Productos Farmacéuticos, Dispositivos Médicos y Productos Sanitarios, luego fue remitido al Presidente de la República para su promulgación, la que fue aprobada y publicada en el Diario Ofcial el Peruano el 26 de noviembre de 2009 (2)
CAMBIO SUSTANCIAL EN LA REGULACIÓN PARA EL REGISTRO SANITARIO
A fin de implementar el Acuerdo de Promoción Comercial suscrito entre el Perú y los Estados Unidos de América (3) se había aprobado la Ley N.º 29316, ley que modifca, incorpora y regula diversas disposiciones (4) , a fn de implementar este acuerdo que modifcó el Artículo 50.º de la Ley General de Salud Ley 26842, relacionado a productos farmacéuticos, galénicos y recursos terapéuticos naturales. Su reglamento, aprobado por Decreto Supremo N.° 001-2009 SA, estableció un cambio sustantivo en la regulación sanitaria de los productos farmacéuticos, cambiando los requisitos sólo para el RS de medicamentos (5) , quedando pendiente otros productos farmacéuticos, como los productos biológicos, medicamentos herbarios, productos dietéticos y edulcorantes y lo afnes referidos a los dispositivos médicos y productos sanitarios. Tampoco contemplaba la regulación de la calidad, el acceso y uso racional de estos productos.
Con la promulgación de la Ley N.º 29459, ley de los productos farmacéuticos, dispositivos médicos y productos sanitarios (2) , se regula a todos estos productos, sustituyendo el Capítulo III del Título II y algunas disposiciones del Capítulo V de la Ley General de Salud. La Ley reconoce los términos armonizados internacionalmente como los dispositivos médicos y productos sanitarios, ya que el término de productos farmacéuticos y afnes había generado a la fecha muchas confusiones y controversias.
Asimismo, se modifcan los aspectos más cuestionados en el Capítulo III de la Ley N.° 26842, al establecer requisitos para el RS de productos farmacéuticos necesarios para garantizar su eficacia, seguridad y calidad.
La Ley N.° 29459, al igual que la Ley N.° 29316, establece criterios para la inscripción y reinscripción de medicamentos, clasifcándolos, para estos efectos, en tres categorías:
Categoría 1. Productos cuyos principios activos o asociaciones se encuentran en el Petitorio Nacional de Medicamentos Esenciales (PNME);
Categoría 2. Productos cuyos principios activos o asociaciones no se encuentran en el PNME y que se encuentran registrados en países de alta vigilancia sanitaria. Dentro de estos se encuentran los productos cuyos principios activos o asociaciones hayan sido registrados en el Perú en la categoría 3 a partir de la vigencia de la ley;
Categoría 3. Productos cuyos principios activos no se encuentran considerados en las categorías 1 y 2.
De acuerdo con cada categoría, se diferencian los requisitos, los cuales garantizan eficacia, seguridad y calidad de los medicamentos, y plazos necesarios para su evaluación, previo al otorgamiento del RS.
REQUISITOS QUE GARANTIZAN LA EFICACIA, SEGURIDAD Y CALIDAD DE LOS MEDICAMENTOS
Es preciso señalar que, mediante el RS de los productos farmacéuticos, dispositivos médicos y productos sanitarios, el Estado avala, certifca y garantiza la seguridad, eficacia y calidad de los productos antes de su ingreso al mercado, evitando así, la existencia en el mercado de medicamentos de valor terapéutico cuestionado.
Para ello, en los artículos 10.° y 11.° de la Ley recientemente promulgada, se establece algunos requisitos para las distintas categorías, en la categoría 1, que corresponde a principios activos o asociaciones Rev Peru Med Exp Salud Publica. 2009; 26(4): 517-29. que se encuentran en el PNME, estos han pasado un proceso de evaluación de seguridad y eficacia en el momento de la selección, entendiéndose que los productos ahí descritos son seguros y efcaces en las formas y concentraciones especifcadas. Este proceso de selección, no es un proceso administrativo como lo consideran muchos profesionales, sino un proceso de investigación, prestando especial atención a su importancia para la salud pública, a las pruebas de seguridad y eficacia (6)
En la categoría 2, sólo se requiere presentar información técnica sobre eficacia y seguridad del principio activo, si es monofármaco, o de la asociación si el producto tiene más de un principio activo; y para los productos de la categoría 3 –que contienen nuevas entidades químicas–, deberán presentar estudios y documentos que sustenten la eficacia y seguridad del producto. La documentación necesaria para realizar una evaluación completa debe contener los datos obtenidos en la investigación preclínica y clínica del nuevo producto farmacéutico y trabajos de desarrollo que condujeron al diseño de cada producto farmacéutico, a efectos de garantizar su eficacia, seguridad y calidad.
En el país, el número de productos nuevos que entran al mercado es pequeño frente al gran número de versiones genéricas de productos ya existentes, con sus nombres de marca y en Denominación Común Internacional (DCI); es importante tener en cuenta que, a la entrada en vigencia de la Ley, miles de estos registros sanitarios existentes van a corresponder a las categorías 2 y 3 y, para esto, se contempla el proceso de reinscripción que la Ley establece: Para la reinscripción de los productos comprendidos en las categorías 2 y 3 que, a la fecha de la vigencia de la Ley cuenten con RS, se presentará adicionalmente información técnica sobre seguridad y eficacia del principio activo o principios activos para el caso de la asociación (2) .
Todo este proceso de evaluación es generalmente complejo, porque ningún medicamento puede considerarse libre de todo riesgo. La evaluación consiste en valorar los riesgos en relación con los benefcios que su uso pueda inducir y, también en las enfermedades prevalentes y las necesidades de salud (7) .
Todos los medicamentos, sean innovadores o genéricos, deben cumplir con los mismos estándares de calidad, eficacia y seguridad. Esto ha sido contemplado en la Ley, ya que en su artículo 10.º para la inscripción y reinscripción del RS, se incluye otros requisitos en las tres categorías como por ejemplo, los estudios de intercambiabilidad, estudios de estabilidad y el certificado de buenas prácticas de manufactura (BPM).
La Organización Mundial de la Salud (OMS), en su informe técnico N.º 937, señala que las autoridades reguladoras de medicamentos deben exigir a los productos genéricos (hoy denominados multifuentes) documentación que asegure que el medicamento reúne los requisitos de BPM, especifcaciones de control de calidad e intercambiabilidad o equivalencia terapéutica con el producto de comparación (8) .
Es preciso señalar que, para que un medicamento genérico sea considerado intercambiable, debe ser terapéuticamente equivalente al producto comparador. Sin embargo, existen productos farmacéuticos para los cuales la intercambiabilidad se asegura por la BPM y especifcaciones, por lo que no requieren estudios de equivalencia terapéutica (8) .
La equivalencia terapéutica, por lo tanto, la intercambiabilidad se puede determinar mediante estudios de bioequivalencia, estudios farmacodinámicos comparativos, estudios clínicos comparativos y estudios in vitro (8-11) . Los avances científcos y tecnológicos en el área de la biofarmacia han permitido desarrollar un sistema de clasifcación biofarmacéutica, según el cual determinados productos farmacéuticos podrán optar por una bioexención, es decir, demostrar su equivalencia terapéutica sin necesidad de realizar estudios comparativos in vivo, sino mediante estudios comparativos in vitro (8) .
Acertadamente la Ley, en su artículo 10.°, plantea, de acuerdo con lo recomendado por la OMS, que los estudios de bioequivalencia (intercambiabilidad) in vivo sean exigibles a los productos de riesgo sanitario alto, considerando las excepciones de acuerdo con la clasifcación biofarmacéutica atendiendo al principio de gradualidad, ello permitirá asegurar la eficacia, seguridad y calidad de los medicamentos que se comercializan en el país.
En tal sentido, para realizar los estudios de equivalencia terapéutica, ya sea a través de estudios comparativos in vivo o a través de estudios in vitro, así como para excluir de estos estudios a determinados productos, se requiere defnir y uniformar las condiciones y requisitos que deberán cumplir los estudios para demostrar equivalencia terapéutica y, por tanto, la intercambiabilidad. Para ello, la DIGEMID se encuentra elaborando, con participación del Centro Nacional de Control de Calidad (CNCC) del Instituto Nacional de Salud (INS), la propuesta de directiva sanitaria que reglamenta los estudios para establecer equivalencia terapéutica de medicamentos, que también cuenta con los aportes y sugerencias de diversas instituciones públicas y privadas.
Otro de los requisitos importantes son los estudios de estabilidad que establece la Ley para los medicamentos comprendidos en las categorías 1, 2 y 3. Mediante estos estudios se garantiza que los productos farmacéuticos que circulan en el mercado reúnan las condiciones de calidad (potencia, identidad, pureza inalterable, etc.), seguridad y eficacia durante su período de vida útil (desde su fabricación hasta el vencimiento declarado por el fabricante), bajo las condiciones de almacenamiento al que es sometido, siendo esto de cumplimiento obligatorio por los laboratorios farmacéuticos, droguerías u otras empresas fabricantes de productos farmacéuticos. Esta información le permitirá proponer el período de validez durante el cual pueda utilizarse el medicamento en forma segura y confable.
La Ley también señala que los estudios de estabilidad deben presentarse de acuerdo con el reglamento aprobado por el MINSA. Por ello es que, mediante la Resolución Ministerial N.º 805-2009/MINSA, se aprueba la directiva sanitaria que reglamenta los estudios de estabilidad de medicamentos (12) .
Por otro lado, hay que tener en cuenta que las sustancias farmacéuticas son biológicamente activas y pueden causar también, en grado variable, efectos indeseables. El riesgo de reacciones graves y de fracaso terapéutico se acentúa cuando los productos son de calidad inferior o se administran incorrectamente. Para evitar ello, la elaboración, envasado y comercialización de productos debe sujetarse a las normas aceptadas internacionalmente, comúnmente conocidas como Buenas Prácticas de Manufactura (BPM) (13) .
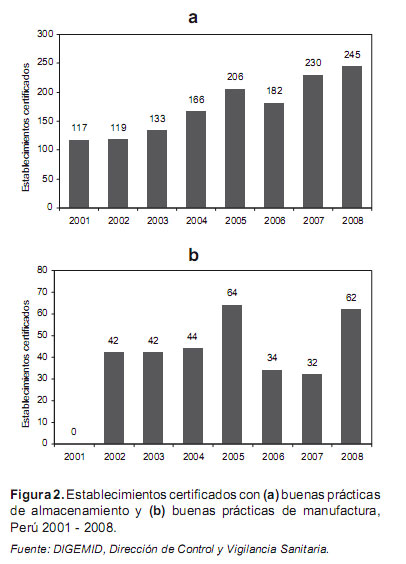
Desde 1997, con la Ley 26842, se estableció la necesidad de que las personas que se dedican a la fabricación o almacenamiento de productos farmacéuticos o ejecuten parte de los procesos, se ciñan a las BPM. De acuerdo con el registro de la DIGEMID, a diciembre de 2008, existen 245 droguerías que cuentan con la certificación de Buenas Prácticas de Almacenamiento –BPA– que representan sólo 6,7% de un total de 3661 registrados y 18,0% (62/345) laboratorios certificado s con BPM (Figuras 2a y 2b).
Por ello, otro de los requisitos importantes para la inscripción y reinscripción del RS que se considera en esta Ley es la presentación del certificado de BPM del fabricante nacional o extranjero emitido por la autoridad nacional de medicamentos (ANM), es decir, la DIGEMID. Esto signifca que la DIGEMID tendrá la autoridad sufciente para inspeccionar plantas de fabricación no solamente de laboratorios nacionales sino también extranjeros y exigir la certificación y su mantenimiento. Así también, hace mención que se aceptará sólo los certifcados de BPM de países de alta vigilancia sanitaria y los países con los que exista reconocimiento mutuo. Todo esto redundará en la calidad de los productos, que estarán respaldados por el cumplimiento de las BPM.
Las BPM constituyen un conjunto de normas mínimas establecidas para la ejecución de los procedimientos destinados a garantizar la calidad uniforme y satisfactoria de los productos farmacéuticos y cuya característica de diseño deben estar dentro de los límites aceptados y vigentes (14) .
La aplicación de las BPM por parte de los fabricantes, asegura que todos los lotes de los productos farmacéuticos sean elaborados con materias primas de calidad adecuada, que cumplan con las especifcaciones de la farmacopea tomada como referencia, que se hayan envasado y rotulado en forma correcta, sean estables y tengan la adecuada disponibilidad durante su vida útil si se mantienen en las condiciones especifcadas en las normas de almacenamiento e indicaciones en el rotulado (15) .
Por otro lado, se faculta al MINSA a defnir las tasas del RS en función de los costos reales, incluyendo adicionalmente los gastos para las acciones de control y vigilancia sanitaria.
La reglamentación modifica también los plazos necesarios para la evaluación y aprobación del RS, categoría 1: 60 días; categoría 2: de 45 a 90 días; y categoría 3: 12 meses.
Es necesario resaltar que, más allá de los beneficios de carácter administrativo que conlleva el proyecto de Ley, existe un beneficio mayor, referido específicamente a la salud de población, ya que se propone un sistema que garantiza condiciones de seguridad, eficacia e idoneidad en cuanto a los productos farmacéuticos, dispositivos médicos y productos sanitarios aptos para ser usado por nuestra población.
SISTEMA DE ASEGURAMIENTO DE LA CALIDAD
La Ley N.° 29459 en su artículo 18.º establece que el control de calidad de los productos farmacéuticos, dispositivos médicos y productos sanitarios es obligatorio, integral y permanente. Para garantizar la calidad de estos productos, los establecimientos públicos y privados, bajo responsabilidad, deben contar con un sistema de aseguramiento de la calidad.
Entendiendo que la calidad involucra todos los aspectos del proceso de fabricación, desde las materias primas empleadas hasta los productos terminados, así como, los procesos de almacenamiento, distribución, dispensación y expendio.
Esto implica establecer un conjunto de acciones planificadas, sistemáticas y objetivas que permitan mejorar la confanza de la ciudadanía en los medicamentos ofertados en el mercado. Para ello, se requiere establecer un sistema de aseguramiento de la calidad que permita interrelacionar cuatro elementos fundamentales:
1. ESTÁNDARES DE CALIDAD
Los estándares de calidad establecidos son la medida sobre la cual las autoridades reguladoras de medicamentos nacional y regional van a desplegar las funciones de control y vigilancia de la calidad de los productos farmacéuticos, dispositivos médicos y productos sanitarios y del buen funcionamiento de los establecimientos farmacéuticos.
Los estándares o requisitos se establecen al momento que se emite el RS de los productos, cuyos requisitos más importantes ya se ha descrito, y la autorización sanitaria de establecimientos farmacéuticos, se realiza previa inspección para verifcar el cumplimiento de los dispositivos legales vigentes, tal como lo señala la ley en su artículo 21.º.
Autorización sanitaria previa al funcionamiento de los establecimientos farmacéuticos
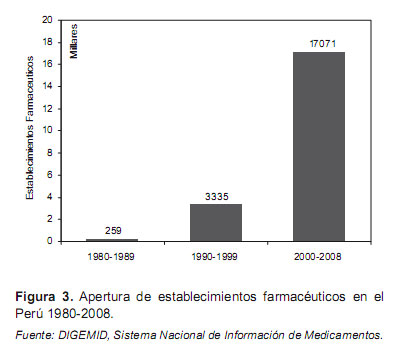
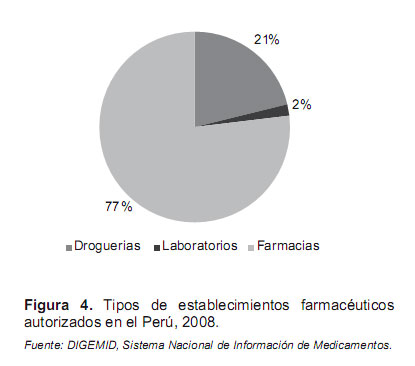
El número de establecimientos farmacéuticos con la legislación anterior, que sólo establecía una simple comunicación con un plazo de 30 días después del inicio de funcionamiento, originó que estos se incrementaran. Al mes de diciembre de 2008 se habían registrado a nivel nacional 13 120 establecimientos entre farmacias y boticas, 3606 droguerías y 345 laboratorios farmacéuticos (Figura 3 y Figura 4).
El control posterior establecido en la anterior Ley no era efectivo, por la simplicidad del trámite administrativo para abrir y cerrar establecimientos, se hacía muy difícil para la ANM la aplicación de sanciones previstas en los reglamentos, ya que inmediatamente después de la inspección realizada en la que se detecta una irregularidad que merecía una multa o el cierre del establecimiento, los propietarios comunican el cierre y la apertura de uno nuevo con alguna modificación de los datos consignados en la primera comunicación tal como el cambio de razón social, del nombre comercial, del propietario, de la dirección u otro. De acuerdo con las acciones de control realizadas, se trata en realidad del mismo establecimiento que continua funcionando con similares defciencias a las detectadas.
La nueva Ley exige ahora, la obligatoriedad de la autorización sanitaria para el funcionamiento de los establecimientos farmacéuticos, previa verifcación del cumplimiento de los dispositivos legales vigentes (2) . La autorización es requisito indispensable para el otorgamiento de las licencia de funcionamiento.
Así también, la Ley establece que habrá un mecanismo para la actualización de la vigencia de la autorización sanitaria, esto signifca que existirá, después de un período, la renovación de la autorización sanitaria, evitando que muchos funcionen de manera irregular a veces largos períodos antes de su detección, lo que implica el consecuente riesgo para la salud.
Las exigencias de calidad que el Estado requiere al solicitante para los establecimientos farmacéuticos, son la obligación de cumplir con las buenas prácticas y contar con la certificación correspondiente.
Antes de esta Ley, sólo se exigía que los establecimientos cumplan con las BPM y BPA, la certificación no era obligatoria; sin embargo, ahora los establecimientos deben cumplir, además, con las Buenas Prácticas de Distribución, Buenas Prácticas de Dispensación y Buenas Prácticas de Seguimiento Farmacoterapéutico.
2. PROCESOS DE CONTROL DE CALIDAD
Son todos los procesos desarrollados para verifcar el cumplimiento de los estándares de los productos farmacéuticos, dispoositivos médicos y productos sanitarios, mediante pesquisas y control de calidad; de los establecimientos farmacéuticos, mediante inspecciones y operativos; de la promoción y publicidad de medicamentos mediante pesquisas/ evaluación. Para la ejecución de estas funciones se debe emplear herramientas de vigilancia y trazabilidad que deberán implementarse gradualmente.
Algunos de estos aspectos ya estaban regulados, sin embargo, existen otros. La nueva Ley (2) en su artículo 45.º contempla requisitos especiales que permitirán un mayor y mejor control de calidad de los productos, hace mención que las empresas deben presentar resultados de control de calidad de todos y cada uno de los lotes que fabrican o importan, con algunas excepciones que se autorizarán por Resolución Ministerial. El control de calidad del primer lote se realiza en el CNCC o los laboratorios autorizados por ellos. Los otros lotes en sus propios laboratorios o por contrato en laboratorios públicos o privados.
Así también, en un proceso de inspección, la ANM tiene la posibilidad de solicitar a la autoridad judicial, la autorización para el ingreso, en caso que no se le permita, actualmente sólo el fscal puede solicitar autorización judicial para ingreso.
¿Por qué son importantes los controles en aduanas de la república?
Anteriormente, las aduanas de la república, para la importación de los productos farmacéuticos y galénicos, exigían únicamente, una declaración jurada consignando lo siguiente: número del RS o, en su defecto, la fecha de presentación de la solicitud correspondiente; identifcación del embarque por lote de producción y fecha del vencimiento del medicamento; tratándose de productos derivados de sangre humana, se exigía un certificado analítico de negatividad de los virus de inmunodefciencia humana y hepatitis virales B y C.
Con la reciente Ley (2) las aduanas exigen requisitos más estrictos como la copia del RS, protocolo de análisis del lote, identifcación del embarque por lote y fecha de vencimiento, certificado BPM del fabricante, entre otros.
Se podrá, ahora, realizar controles en coordinación con las aduanas del país. Para el control de calidad de los productos en aduana, se podrá hacer las verifcaciones o pesquisas en la zona primaria aduanera (durante el reconocimiento físico o después del levante autorizado) y en los almacenes aduaneros, previa autorización de la autoridad aduanera.
Asimismo, los almacenes aduaneros para productos farmacéuticos, dispositivos médicos y productos sanitarios deben estar acondicionados con la BPA. Anteriormente este acondicionamiento de las BPA, sólo era para establecimientos farmacéuticos que almacenan, no depósitos aduaneros ni receptores de donaciones.
Comercio ilegal de medicamentos
La OMS hace referencia sobre la magnitud del problema, aunque es difícil obtener cifras precisas, se calcula que los medicamentos falsifcados representan más de 10% del mercado farmacéutico mundial (16) . Si bien esta práctica existe en todas las regiones, en los países en desarrollo se estima que 25% de los medicamentos que se consume son falsifcados (17) , convirtiéndose esto en un grave problema de salud pública que afecta a la población, a las empresas formales y al Estado.
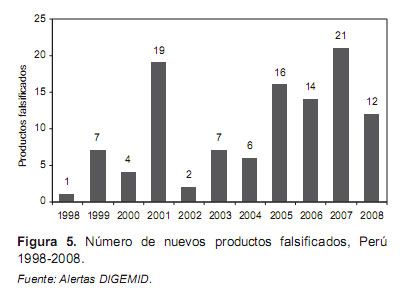
Entre las estrategias y acciones orientadas a combatir el comercio ilegal de los productos farmacéuticos y afnes, destacaban entre otros: la verifcación in situ de las observaciones señaladas en las denuncias, la verifcación física organoléptica de los productos presuntamente falsifcados, remitidos por las direcciones regionales de salud (DISA/DIRESA), además de realizar operativos a establecimientos formales e informales y la evaluación de los productos farmacéuticos y afnes incautados y decomisados por otras instituciones.
Durante el período 2004-2008 se evaluó 1 773 176 unidades incautadas o decomisadas a nivel nacional; remitidos a la DIGEMID a fn de elaborar los informes técnicos, los cuales servirán de sustento para realizar las denuncias penales correspondientes.
En la Figura 5, se observa que durante el período 19972008, producto de los operativos realizados a nivel nacional, se encontró 109 nuevos productos farmacéuticos falsifcados en diversas formas farmacéuticas, siendo las más falsifcadas la forma farmacéutica de tabletas e inyectables.
Mediante RM N.º 047-2006-PCM se conformó el Grupo Técnico Multisectorial de Prevención y Combate al Contrabando, Comercio Informal y Falsifcación de Productos Farmacéuticos y Afnes (CONTRAFALME), con el fn de fortalecer estas acciones, integrado por diversas instituciones públicas y privadas, liderada por el MINSA a través de la DIGEMID. La normatividad de este grupo técnico ahora tiene rango de Ley.
Promoción y publicidad farmacéutica
Desde muchos años atrás, la OMS ha advertido sobre los efectos negativos que puede tener la promoción farmacéutica sobre el uso de los medicamentos. En 1968 la Asamblea Mundial de la Salud aprobó los criterios éticos para la promoción de medicamentos.
En el Perú, sin embargo, estos criterios están considerados dentro del Reglamento de la Ley General de Salud (18) , pero sólo aplicados a la promoción y publicidad de productos farmacéuticos y productos naturales cuya condición de venta sea sin receta médica. La nueva Ley establece que la información difundida con fnes de promoción y publicidad de productos farmacéuticos, dispositivos médicos y productos sanitarios, debe ser concordante con lo autorizado en el registro sanitario y sujetarse a los criterios éticos para la promoción de medicamentos establecidos por la autoridad nacional de salud.
En el año 2007, la Asamblea Mundial de la Salud reconoció la gran infuencia de la promoción farmacéutica en la calidad del uso de los medicamentos y recomienda a los países miembros de la organización a que promulguen nuevos textos legislativos, o los hagan cumplir cuando ya existan, que prohíban la promoción inexacta, equívoca o no ética de medicamentos, a que vigilen la promoción de medicamentos y a que elaboren y apliquen programas para ofrecer información independiente y no promocional sobre los medicamentos (19)
Para inducir al consumo de sus productos, los fabricantes y distribuidores utilizan variados mecanismos de persuasión sobre los actores que seleccionan, adquieren, prescriben y usan los medicamentos (20,21) .
Durante el año 2008, el número de anuncios publicitarios evaluados fueron 1656, de los cuales, 47,6% corresponde a productos para venta con receta médica y 52,4% (867) a productos para venta sin receta médica, dentro de los cuales tenemos: medicamentos, productos naturales, dietéticos, cosméticos, productos de higiene sanitaria, material e instrumental médico entre otros.
Es el INDECOPI quien supervisa y aplica sanciones en materia publicitaria, y en este período se efectuó 38 denuncias por transgresiones a las normas vigentes. El INDECOPI inició procedimiento de ofcio a 8 (21%) de las denuncias presentadas por la DIGEMID (22) .
Entre las prohibiciones que se agregan a las ya existentes se encuentra cualquier actividad que incentive la venta, prescripción o dispensación de productos farmacéuticos en los establecimientos farmacéuticos de dispensación.
Esto es importante porque las prohibiciones ya no sólo se limitan a los productos de venta con receta médica sino también a los de venta sin receta médica. Es importante saber que todo medicamento es un tóxico, pero que usado de manera adecuada puede ser muy beneficioso para la salud. Si bien existen ciertos medicamentos que por su baja toxicidad se autorizan para la venta sin receta médica, eso no implica que puedan ser distribuidos indiscriminadamente en calles, plazas, playas, centros comerciales u otros, en donde muchas veces pueden ser recibidos por personas que les pueden dar un uso inadecuado. Se presentan casos en los cuales se distribuyen medicamentos a niños en los colegios o se hacen degustaciones en establecimientos farmacéuticos y otros, sin tener en consideración que los medicamentos sólo deben ser usados cuando son necesarios.
La Autoridad Nacional de Salud regula la promoción médica en establecimientos de salud, así como la entrega directa al público de muestras con receta médica por las empresas farmacéuticas, visitadores médicos o promotores; estos aspectos no estaban regulados.
De igual manera, permite el establecimiento de mecanismos de coordinación con el INDECOPI en temas relacionados con marcas de implicancia sanitaria. Y espacios gratuitos en medios de comunicación del Estado para la difusión de información sobre productos farmacéuticos, no había regulación sobre espacios gratuitos
3. MEJORA CONTINUA DE LA CALIDAD
Considera los procesos para lograr una mejora continua de la calidad de los medicamentos y el buen funcionamiento de los establecimientos farmacéuticos, a través de la certificación de sus sistemas de calidad y verifcando las debidas competencias a las DIRESA y las empresas privadas. Esto lo señala la Ley (2) en su artículo 22.° al referirse que la autoridad de medicamentos a nivel nacional es la encargada de otorgar la certificación de las buenas prácticas a los establecimientos farmacéuticos, ello con la finalidad de garantizar la calidad de los establecimientos públicos y privados.
Así mismo, previa verifcación de las condiciones necesarias, transferirá las funciones de certificación a los órganos desconcentrados y a la autoridad regional de medicamentos.
4. RELACIÓN PROVEEDOR – USUARIO
Tiene que ver con todos los procesos desarrollados para lograr una interrelación entre el sistema y el usuario, que implica la retroalimentación necesaria para el desarrollo y mejora continúa del sistema.
La DIGEMID emplea como inputs, las denuncias de los usuarios, quejas y reclamos, mediciones de satisfacción de cliente, acciones de farmacovigilancia y tecnovigilancia, mesas de trabajo con actores relevantes del sector salud, entre otros. Los outputs vienen a ser los servicios y productos por lograr, como las alertas de calidad, observatorio de calidad y precios, los procesos de educación, capacitación, sensibilización de la comunidad y las acciones de interrelación con los actores relevantes del sector salud entre otros.
Para esto y, cumpliendo un anhelo largamente esperado y como resultado de un dedicado trabajo que consistió en la implementación de un sistema de gestión de la calidad, que representa la institucionalización del mejoramiento continuo en cada uno de los procesos que desarrolla, la DIGEMID obtuvo de parte de la empresa internacional ICONTEC la certificación del ISO 9001:2000.
El proceso de implementación del ISO 9001:2000 se inició con los procesos relacionados con la inscripción y reinscripción de productos farmacéuticos y afnes; fjándose, cada vez, nuevas y mayores metas y, por supuesto, ampliar el alcance del sistema a todas las áreas de la DIGEMID. Para ello se propuso como política de la calidad:
-
Desarrollar sus actividades buscando brindar un mejor servicio a los clientes.
-
Aplicar la mejora continua en cada uno de sus procesos.
-
Establecer y mantener un sistema de gestión de la calidad basado en el cumplimiento de la norma ISO 9001-2001 y la legislación vigente.
-
Proporcionar a los trabajadores capacitación y recursos necesarios para lograr los objetivos trazados.
Es importante señalar que la Ley prevé en su artículo 24.° que la ANM publicará el listado de establecimientos autorizados, señalando la condición en que se encuentren. Esto permitirá que la población se mantenga informada acerca de los establecimientos que reúnen las condiciones sanitarias.
Asimismo, la ANM, establece y pública el estándar único de identifcación de productos farmacéuticos, dispositivos médicos y productos sanitarios, para dar trazabilidad al producto que colocan en el mercado.
La DIGEMID. está trabajando en la implementación del Observatorio de Precios, con la fnalidad de informar a la población sobre el comportamiento de los precios de venta de los medicamentos esenciales en establecimientos públicos y privados, utilizar información actualizada de los precios de adquisición de medicamentos existentes en el mercado para determinar el Valor Referencial de los Procesos de Selección que convoquen las entidades del sector público nacional y vigilar el impacto que genera en los precios de los medicamentos.
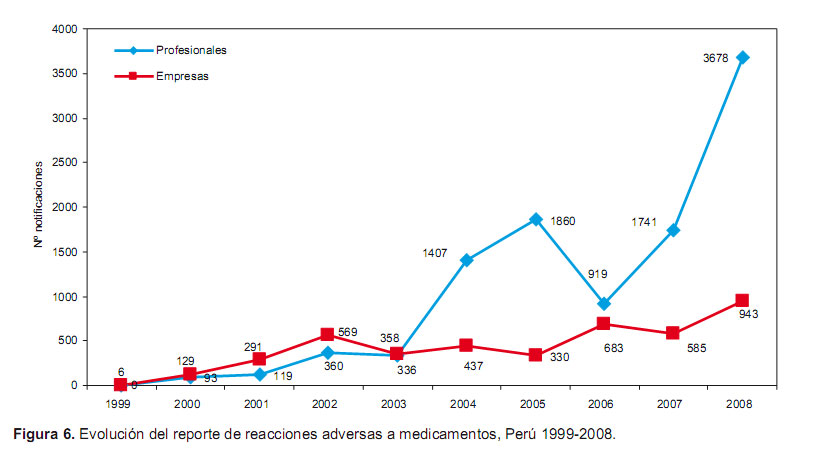
Otra de las acciones es la farmacovigilancia, la cual es una actividad de salud pública que se encarga de la detección, la evaluación y la prevención de los riesgos asociados con los medicamentos una vez comercializados. La DIGEMID en el año 1999 aprobó el Sistema Peruano de Farmacovigilancia (SPFV) y, desde el año 2002, es uno de los miembros activos del Programa Internacional de Farmacovigilancia de la OMS.
Es decir, ya tiene 10 años en funcionamiento, el crecimiento del sistema, medido en función al número de reportes, fue progresivo, tal como se demuestra en la Figura 6. Sin embargo, todavía se encuentra en un nivel bajo de notifcación, dada la cantidad de medicamentos que circulan en el mercado y el número de profesionales de la salud, para ello se continúa promoviendo la farmacovigilancia entre los profesionales de la salud.
La DIGEMID ha venido trabajando y recibiendo reportes de eventos adversos por el uso de dispositivos médicos, sin embargo, no contábamos con una base legal que lo dispusiera, con esta Ley, en su artículo 35.° ya nos da la oficialidad del Sistema Peruano de la Tecnovigilancia, que se encarga de la prevención, detección, investigación, evaluación y difusión de información sobre incidentes adversos o potencialmente adversos relacionados con dispositivos médicos durante su uso; y, en su artículo 36.° la obligación de reportar las sospechas de reacciones y eventos adversos de los medicamentos, otros productos farmacéuticos, dispositivos médicos y productos sanitarios que prescriben , dispensan o administran.
DEL ACCESO Y USO RACIONAL DE PRODUCTOS FARMACÉUTICOS, DISPOSITIVOS MÉDICOS Y PRODUCTOS SANITARIOS
Es importante resaltar que en esta nueva Ley se incorporan tres capítulos específcos sobre acceso, uso racional de productos farmacéuticos, dispositivos médicos y productos sanitarios, así también, un capítulo de investigación. La anterior sólo resaltaba estos temas y los resumía en un sólo artículo (el 75.º).
Asimismo, la Ley recoge dos de sus lineamientos básicos que están establecidos en la Política Nacional de Medicamentos, aprobada por Resolución Ministerial N.º 1240-2004/MINSA, como son: el acceso universal a los medicamentos, con el objetivo especifco de asegurar el acceso universal a medicamentos esenciales como componente fundamental de la atención integral en salud y la Promoción del Uso Racional de Medicamentos con el objetivo de fomentar esta cultura a nivel nacional (23) .
La Ley también incluye los fundamentos básicos del acceso universal: selección racional, promoción y fortalecimiento de la fabricación y prescripción de medicamentos genéricos, precios asequibles, transparencia de la información, suministro efciente y oportuno, fomentar sistemas de dispensación de medicamentos en dosis unitarias, nutrición artifcial, mezclas intravenosas y atención farmacéutica en establecimientos de salud a nivel nacional, fomentar la investigación y las medidas de aseguramiento universal.
La Ley también da la facultad de aplicar las limitaciones y excepciones previstas en el acuerdo sobre los aspectos de los derechos de propiedad intelectual relacionados con el comercio (ADPIC), sus enmiendas y la Declaración de DOHA.
El acceso a los medicamentos es reconocido como un componente esencial para el pleno ejercicio del derecho a la salud. El derecho de las personas a acceder a los servicios de salud en general y a los medicamentos en particular, exige tener en cuenta a) accesibilidad geográfca; b) disponibilidad, referida a que los servicios de dispensación y expendio de medicamentos deben existir permanentemente c) aceptabilidad de los servicios de salud, se refere a que éstos deben tomar en cuenta las culturas de las poblaciones y sus percepciones sobre salud-enfermedad d) asequibilidad, referida a que los servicios y medicamentos deben estar al alcance de las economías de las personas y las comunidades.
La pobreza, educación y salud son factores estrechamente ligados que infuyen sobre el acceso a medicamentos e insumos esenciales, particularmente para los sectores menos favorecidos y para quienes se debe especialmente organizar los servicios del sector público.
El MINSA, a través del Sistema Integrado de Suministro de Medicamentos e Insumos Medico-Quirúrgicos (SISMED) (24,25) ha avanzado signifcativamente en la búsqueda de nuevas modalidades de compra de medicamentos. Entre los años 2003 y 2005, el MINSA/ DIGEMID efectuó procesos de compra corporativa de medicamentos para todas sus unidades ejecutoras a nivel nacional, desde el año 2006 a la fecha, viene ampliando su capacidad de compra corporativa facultativa intersectorial (economía de escala) y su liderazgo en todo el sector público (MINSA, ESSALUD, Ministerio de Defensa y Ministerio del Interior). Aplicando la modalidad de subasta inversa, mejorando la efciencia, reportando signifcativos ahorros públicos por reducción sustancial de los precios, disponibilidad oportuna, garantizando la calidad previamente a su distribución a nivel nacional, permitiendo que mayor población pueda acceder a ellos, sobre todo a los que no tienen ningún tipo de aseguramiento.
Las compras nacionales están llegando en los últimos años a alrededor de 125 millones de nuevos soles. En los tres procesos de compras, se logró alcanzar ahorros por el equivalente a S/. 128 694 358 nuevos soles.
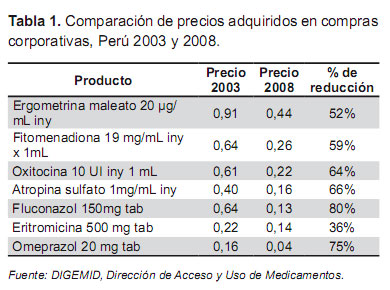
En la Tabla 1 se puede observar la reducción sustancial de precios hasta en 75%, de medicamentos comunes que se ofertan en los establecimientos de salud del MINSA.
Según la OMS, el uso racional de los medicamentos requiere que Los pacientes reciban la medicación adecuada a sus necesidades clínicas, en las dosis correspondientes a sus requisitos individuales, durante un período adecuado y al menor costo posible para ellos y para la comunidad (26) .
En este contexto y en el marco de la Política Nacional de Medicamentos (23) , la DIGEMID viene realizando acciones orientadas a fomentar una cultura de uso racional de los medicamentos a nivel nacional, acciones que incluyen, entre otras, la elaboración del petitorio nacional único de medicamentos esenciales para el sector público, esto a través del grupo de trabajo multisectorial creado por resolución ministerial N.° 007-2008 PCM que asumió la responsabilidad de elaborar la propuesta, aspecto que también ha sido contemplado por la actual Ley (2) en su artículo 34.º.
El petitorio nacional único de medicamentos esenciales para el sector público se elabora mediante un proceso de selección de los medicamentos esenciales porque ayuda a establecer prioridades dentro de un sistema sanitario y permite garantizar la atención de los problemas prioritarios de salud de la población.
El PNME, vigente a la fecha, incluye 365 principios activos en 577 presentaciones farmacéuticas, sólo tiene alcance para las instituciones del MINSA Salud, pero no para otras dependencias públicas; la segmentación de la prestación de servicios ha originado la existencia de petitorios de medicamentos entre diversos órganos estatales: Ministerio de Defensa (Fuerza Aérea, Marina de Guerra y Ejército), Ministerio del Interior, Ministerio de Salud, ESSALUD con diversidad de principios activos.
La irracionalidad en el uso de medicamentos, así como la prescripción y dispensación inapropiadas se ven condicionadas por la defciente formación profesional, así como, por la promoción y publicidad farmacéutica sesgada, que junto con la automedicación no informada, constituyen una realidad que se debe modificar (23) .
Por ello, es importante que en el capítulo del uso racional en su artículo 31.º hace mención que la prescripción de medicamentos debe hacerse consignando obligatoriamente la DCI, teniendo en consideración lo establecido en las buenas prácticas de prescripción. Se debe tener en cuenta que en una buena prescripción buscamos maximizar la efectividad en el uso de los medicamentos, minimizar los riesgos a los que se somete el paciente al usar un medicamento, minimizar los costos en la atención de salud por medio del uso racional del medicamento.
Así también, en el artículo 32.º se dispone que la dispensación de los productos comprendidos en la Ley, debe hacerse siguiendo lo normado en las buenas prácticas de dispensación y seguimiento fármaco terapéutico, las cuales constituyen la atención farmacéutica.
Finalmente, mencionar que La Ley recoge varias líneas estratégicas de la Política Andina de Medicamentos (27) elaborada por la Comisión Técnica Subregional para la Política de Acceso a Medicamentos, con el apoyo de la Organización Panamericana de la Salud (OPS) y la Cooperación Francesa, con el propósito de armonizar políticas y diseñar estrategias para enfrentar problemas comunes de los países andinos, orientados a fortalecer la gestión sanitaria del medicamento y diseñar acciones conjuntas dirigidas a lograr que la población de la subregión andina cuente con medicamentos efcaces, seguros y de calidad, promoviendo su uso racional y garantizando acceso equitativo a aquellos que son esenciales. Esto representa un hecho de singular valor, ya que defne una política común para un área de gran importancia estratégica. La PAM se construyó sobre cuatro grandes líneas estratégicas: acceso universal; calidad, eficacia y seguridad; uso racional e investigación y desarrollo.
Esta política fue aprobada por los ministros de Bolivia, Chile, Colombia, Ecuador, Perú y Venezuela en la XXX Reunión de Ministras y Ministros de Salud del Área Andina (REMSAA), celebrada en Lima en marzo de 2009.
Conflictos de interés
El autor es Director General de la Dirección General de Medicamentos, Insumos y Drogas del Ministerio de Salud del Perú.
REFERENCIAS BIBLIOGRÁFICAS:
1. Perú, Congreso de la República. Ley Nº 26842: Ley general de salud. Lima: Congreso de la República; 1997
2. Perú, Congreso de la República. Ley Nº 29459: Ley de los productos farmacéuticos, dispositivos médicos y productos sanitarios, se regula a todos los productos farmacéuticos, dispositivos médicos y productos sanitarios. Lima: Congreso de la República; 2009.
3. Perú, Congreso de la República. Ley Nº 29157: Ley que delega en el Poder Ejecutivo la facultad de legislar sobre diversas materias relacionadas con la Implementación del Acuerdo de Promoción Comercial PerúEstados Unidos, y con el apoyo a la competitividad económica para su aprovechamiento. Lima: Congreso de la República; 2008.
4. Perú, Congreso de la República. Ley Nº 29316: Ley que modifica, incorpora y regula diversas disposiciones a fin de implementar El Acuerdo de Promoción Comercial Suscrito Entre el Perú y los Estados Unidos. Lima: Congreso de la República; 2009.
5. Perú, Ministerio de Salud. Decreto Supremo N° 0012009-SA: Reglamento del artículo 50° de la Ley N° 26842, Ley General de Salud. Lima: MINSA; 2009
6. Perú, Ministerio de Salud, Dirección General de Medicamentos, Insumos y Drogas. Manual de selección de medicamentos esenciales, principios para una selección racional de medicamentos. Lima: MINSA/DIGEMID; 2001.
7. Vernengo M. Elementos técnicos de una política de medicamentos genéricos. Washington DC: Organización Panamericana de la Salud; 1993.
8. World Health Organization. WHO Expert Committee on Specifications for Pharmaceutical Preparations, Fortieth Report. Geneva: World Health Organization, 2006. WHO Technical Report Series N.º 937.
9. Red Panamericana para la Armonización de la Reglamentación Farmacéutica, Grupo de Trabajo en Bioequivalencia. Marco para la ejecución de los requisitos de equivalencia para los productos farmacéuticos. Washington DC: OPS; 2008.
10. The European Agency for the Evaluation of Medicinal Products, Committee for Proprietary Medicinal Products. Note for guidance on the investigation of bioavailability and bioequivalence. London: EMEA; 2000.
11. The European Agency for the Evaluation of Medicinal Products, Committee for Medicinal Products for Human Use. Guideline on the investigation of bioequivalence. London: EMEA; 2008.
12. Perú, Ministerio de Salud, Dirección General de Medicamentos, Insumos y Drogas. Directiva sanitaria que reglamenta los estudios de estabilidad de medicamentos. Lima: MINSA/DIGEMID; 2009.
13. World Health Organization. WHO Expert Committee on Specifications for Pharmaceutical Preparations, Thirtyseventh Report. Geneva: World Health Organization, 2003. WHO Technical Report Series N.º 908.
14. Perú, Ministerio de Salud. Decreto Supremo N° 0212001-SA: Reglamento de establecimientos farmacéuticos. Lima: MINSA; 2001.
15. Perú, Ministerio de Salud, Dirección General de Medicamentos, Insumos y Drogas. Manual de buenas prácticas de manufactura de productos farmacéuticos. Lima; MINSA/DIGEMID; 1999.
16. Organización Mundial de la Salud. 62ª Asamblea Mundial de la Salud A62/13: Falsificación de productos médicos. Ginebra: OMS; 2009.
17. Morris J, Stevens P. La falsificación de medicamentos en los países menos desarrollados: problemas y soluciones. Londres: International Policy Networks; 2006.
18. Perú, Ministerio de Salud. Decreto Supremo N° 010-97 SA: Reglamento para el registro, control y vigilancia sanitaria de productos farmacéuticos y afines. Lima: MINSA; 1997.
19. Organización Mundial de la Salud. 60ª Asamblea Mundial de la Salud WHA60.16: Progresos realizados en el uso racional de los medicamentos. Ginebra: OMS; 2007.
20. Gagnon MA, Lexchin J. The cost of pushing pills: a new estimate of pharmaceutical promotion expenditures in the United States. PLoS Med. 2008; 5(1): e1.
21. Mitchell PB. Winds of change: growing demands for transparency in the relationship between doctors and the pharmaceutical industry. Med J Aust. 2009; 191(5): 273-75.
22. Perú, Presidencia de la República. Decreto Legislativo Nº 1044: Ley de represión de la competencia desleal. Lima: Congreso de la República; 2008.
23. Perú, Ministerio de Salud, Dirección General de Medicamentos, Insumos y Drogas. Política Nacional de Medicamentos. Lima: MINSA/DIGEMID; 2004.
24. Perú, Ministerio de Salud, Dirección General de Medicamentos, Insumos y Drogas. Directiva del Sistema Integrado de Suministro de Medicamentos e Insumos Médico Quirúrgicos (SISMED). Lima: MINSA/DIGEMID; 2002.
25. Perú, Ministerio de Salud, Dirección General de Medicamentos, Insumos y Drogas. Modificatoria de la Directiva del Sistema Integrado de Suministro de Medicamentos e Insumos Médico Quirúrgicos (SISMED). Lima: MINSA/ DIGEMID; 2005.
26. Conference of Experts on the Rational Use of Drugs. The rational use of drugs. Report of the Conference of Experts, Nairobi 25-29 Nov, 1985. Geneva: World Health Organization; 1987.
27. Organismo Andino de Salud – Convenio Hipólito Unanue. Política andina de medicamentos. Lima: ORASCONHU; 2009.
Correspondencia: Dr. Victor Dongo Zegarra
Dirección: Calle Coronel Odriozola 111 San Isidro, Lima, Perú.
Teléfono: (511) 422-9200
Correo electrónico: vdongoz@yahoo.es
Recibido: 09-12-09
Aprobado: 09-12-09

