










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
versión impresa ISSN 1726-4634
Rev. perú. med. exp. salud publica v.26 n.4 Lima oct./dic. 2009
SIMPOSIO: POLÍTICA DE MEDICAMENTOS
Estudios de bioequivalencia: la necesidad de establecer la fiabilidad de los medicamentos genéricos
Bioequivalence studies: need for the reability of generic drugs
Olga Laosa1,a, Pedro Guerra1,a, José Luis López-Durán1,a, Beatriz Mosquera1,b, Jesús Frías1,2,a
1 Centro de Farmacología Clínica, Departamento de Farmacología y Terapéutica, Facultad de Medicina, Universidad Autónoma de Madrid. Madrid, España.
2 Servicio de Farmacología Clínica, Hospital Universitario la Paz. Madrid, España.
a Médico especialista en Farmacología Clínica; b Licenciada en Ciencias Químicas.
RESUMEN
Un medicamento genérico es un medicamento que contiene un principio activo ya conocido y previamente desarrollado e inventado por otros. El coste de estos productos genéricos o multifuente debe ser menor que el de sus contrapartidas originales. Los efectos clínicos y el balance riesgo-beneficio de un medicamento no dependen exclusivamente de la actividad farmacológica de la sustancia activa. La demostración de bioequivalencia de los medicamentos genéricos es de gran importancia. En Europa y en los Estados Unidos de Norteamérica la autorización de medicamentos genéricos descansa en la demostración de la bioequivalencia mediante estudios de biodisponibilidad comparada in vivo. Estos argumentos son imprescindibles para la autorización de la comercialización de los fármacos genéricos por parte de las autoridades sanitarias europeas y norteamericanas. Como medida de la cantidad de fármaco absorbido se utiliza el área bajo la curva concentración-tiempo (AUC), y como indicador de la velocidad de absorción se mide la concentración máxima (Cmax) alcanzada en la curva concentración-tiempo y el tiempo que tarda en alcanzarse (Tmax). Se entiende por bioequivalencia entre dos productos cuando presentan una biodisponibilidad comparable en condiciones experimentales apropiadas. El objetivo final de todo este proceso tiene como único sentido poner a disposición de la sociedad fármacos de calidad, que además puedan contribuir a una utilización más racional de los recursos económicos en el sistema sanitario.
Palabras clave: Medicamentos genéricos; Política de medicamentos genéricos; Equivalencia terapéutica; Ensayos clínicos como asunto; Área bajo la curva (fuente: DeCS BIREME).
ABSTRACT
A generic medicine is a pharmaceutical product containing an active ingredient already known and previously developed and invented by others. The cost of these generic or multisource products should be less than their counterparts original. The clinical effects and the risk-benefit balance of a medicine do not depend exclusively on the activity of a pharmacologically active substance. Demonstration of bioequivalence of generic medicine is of great importance. In Europe and the United States generic medicine approval is based in the demonstration of bioequivalence through comparative bioavailability studies in vivo. These arguments are required for marketing approval of generic medicines by the European and North American health authorities. As a measure of the amount of drug absorbed it is used the area under the curve concentrationtime (AUC), and as an indicator of the rate of absorption it is measured the peak concentration (Cmax) reached in the concentration-time curve and the time for its occurrence (Tmax). It is known as bioequivalence between two products when they have a comparable bioavailability in the appropriate experimental conditions. The ultimate goal of this process is to make quality drugs available to society and contribute to a more rational use of economic resources in the health system.
Key words: Generic drugs; Generic drug policy; Therapeutic equivalence; Clinical trials as topic; Area under curve (source: MeSH NLM).
INTRODUCCIÓN
El incremento en los costos de los medicamentos se ha convertido en un aspecto trascendente en la política sanitaria en muchos países, comprometiendo los presupuestos de pacientes particulares, de las aseguradoras proveedoras de medicamentos e incluso de los propios sistemas nacionales de salud. Uno de los mecanismos potencialmente capaces de limitar el gasto farmacéutico es la convivencia en el mercado farmacéutico de medicamentos originales o innovadores, protegidos o no por patente, con otros medicamentos llamados multifuente o genéricos, que suelen tener un menor costo que el de los originales durante el periodo de protección de patente o de exclusividad en el mercado. Muchos proveedores de medicamentos han favorecido la presencia en el mercado de medicamentos genéricos con el objetivo de abaratar costos sustituyendo a los medicamentos originales, habitualmente más caros, incluso hasta el punto de que en algunos países los sistemas públicos de salud han implementado diferentes mecanismos, como la sustitución por genéricos, en orden a controlar la creciente factura del gasto farmacéutico.
El realidad, un medicamento genérico es un medicamento que contiene un principio activo ya conocido y previamente desarrollado e inventado por otros. El costo de este tipo de productos debe ser menor que el de sus contrapartidas originales porque su desarrollo y comercialización es mucho más sencilla, puesto que no tiene que demostrar su eficacia y seguridad en largos y costosos ensayos clínicos, dado que ha sido bien establecida por el innovador y por el uso continuado en la práctica clínica. De hecho, los fabricantes de estos medicamentos genéricos sólo tienen que demostrar que su formulación contiene el mismo principio activo que el innovador y que se comporta en el organismo de la misma manera, es decir de manera equivalente.
Sin embargo, estos medicamentos genéricos son equivalentes farmacéuticos con la marca registrada original en términos de ingredientes activos, pero pueden diferir en otros componentes como los saborizantes, estabilizadores, y demás excipientes, además del proceso de fabricación y en el propio laboratorio fabricante. Y ello es muy importante, porque es bien sabido que los efectos clínicos y el balance riesgo-beneficio de un medicamento no dependen exclusivamente de la actividad farmacológica de la sustancia activa sino que también influye la farmacocinética y la forma de acceder el medicamento al organismo.
Así, la demostración de bioequivalencia de los medicamentos genéricos es de gran importancia porque, teóricamente, cualquier medicamento genérico debe ser bioequivalente con su contraparte registrada y por tanto podría ser intercambiado.
En muchos países el propósito fundamental de las agencias regulatorias es garantizar una regulación sobre fabricación y distribución de productos farmacéuticos para uso humano que permita la salvaguarda de la salud pública. En estos países el proceso de autorización de los productos genéricos está claramente establecido y descansa en la demostración de la bioequivalencia, definida como ausencia de diferencias significativas en la biodisponibilidad del ingrediente activo en el lugar de acción del fármaco.
Una vez admitida la necesidad de demostrar la equivalencia como un aspecto relevante que permita establecer la relación entre el producto genérico y el innovador que demostró eficacia y seguridad, se convierte en trascendente la forma de establecer esta equivalencia. Diferentes expertos y autoridades han propuesto diversas formas, entre otras, por ejemplo, la realización de pruebas de disolución in vitro, ensayos clínicos comparativos, estudios farmacodinámicos o estudios de biodisponibilidad comparada in vivo.
La Unión Europea (UE) ha establecido que un producto farmacéutico genérico es un producto medicinal que tiene la misma composición cualitativa y cuantitativa en sustancia activa, la misma forma farmacéutica que el medicamento de referencia y cuya bioequivalencia haya sido demostrada por estudios de biodisponibilidad adecuados (1,2). En esta definición se asume que, en un mismo sujeto, un curso temporal de concentraciones plasmáticas similares resultarán en concentraciones esencialmente iguales en el lugar de acción y por tanto, un efecto básicamente igual. Por tanto, se podrían utilizar datos farmacocinéticos en lugar de datos clínicos para establecer la equivalencia terapéutica, es decir, la bioequivalencia. De esta manera, en Estados Unidos de Norteamérica (EEUU) y en Europa la forma clásica de garantizar la equivalencia entre formulaciones diferentes es la así llamada bioequivalencia (equivalencia in vivo) basada en la comparación de la biodisponibilidad in vivo usando parámetros farmacocinéticos.
Otros países utilizan el término y el concepto de medicamento genérico de diversa manera, por ejemplo para referirse a medicamentos copia de otros, aunque no se haya extinguido el periodo de protección de patente, igualmente han incorporado normativas sobre bioequivalencia con distintos requisitos de acuerdo con cada contexto nacional específico y dependiendo de las características de las sustancias activas, de los riesgos para la salud o de la disponibilidad de recursos para desarrollar estudios más exigentes. En muchos países existen intereses para establecer definiciones y regulación respecto de los estudios de bioequivalencia in vivo y para la aceptabilidad de estudios in vitro. Incluso en EEUU y en Europa existe la posibilidad de solicitar, para un número limitado de medicamentos, autorizaciones de productos genéricos basadas en la demostración de la equivalencia mediante pruebas in vitro (bioexención), pero la metodología de estas pruebas es más exigente que la de los habituales controles de calidad y el sistema continua siendo excepcional (3), de manera que en esas latitudes la autorización de me- dicamentos genéricos descansa en la demostración de la bioequivalencia mediante estudios de biodisponibilidad comparada in vivo.
En Europa y en EEUU sólo se pueden comercializar medicamentos genéricos una vez que se han extinguido los derechos de patente, requiriéndose, además, una evaluación técnica y administrativa que garantice que se cumplen las exigencias establecidas por la ley sobre calidad, seguridad y eficacia. El perfil de seguridad y eficacia debe estar bien establecido y acreditado por su uso clínico, es decir por la experiencia que permite un uso clínico prolongado, mientras que los requisitos de calidad exigidos para los genéricos son exactamente los mismos que para cualquier otro medicamento, definiéndose mediante el grado con que es capaz de reproducir y mantener sus características de seguridad y eficacia a lo largo de todas sus fabricaciones y durante su vida de utilización (4).
Los estudios que avalan la calidad de un medicamento genérico, se recogen en la documentación química, farmacéutica y biológica del dossier. En el caso de medicamentos genéricos, no es necesaria la presentación de estudios toxicológicos y de eficacia, pero sí se debe desarrollar los estudios de estabilidad del preparado, tanto a largo plazo y tiempo real, para conocer el plazo de validez en el que se garantiza que se mantienen las condiciones de seguridad y eficacia del producto, como en condiciones aceleradas, para conocer las condiciones especiales que precisa el preparado. A medida que se van fabricando estos preparados se debe estudiar también la similitud con el preparado de referencia mediante la prueba de disolución in vitro bajo determinadas condiciones, y cuando se aprecia suficiente similitud se aborda la prueba definitiva que es el estudio de bioequivalencia in vivo.
ESTUDIOS DE BIOEQUIVALENCIA
GENERALIDADES
Durante el desarrollo clínico de los medicamentos es frecuente que las formas farmacéuticas que se utilizan en las fases iniciales de la investigación de la eficacia y seguridad de los fármacos en seres humanos sean distintas de las que se utilizarán en las fases posteriores del desarrollo clínico o de las que se emplearán una vez que el fármaco haya sido aprobado para su comercialización. De hecho, alrededor del 60% de las formas farmacéuticas que se utilizan una vez que los fármacos están autorizados para su comercialización son diferentes de las probadas inicialmente. Este cambio en las formas farmacéuticas hace necesario disponer de estudios farmacocinéticos que describan y comparen la biodisponibilidad, esto es la velocidad y la cantidad de fármaco que el organismo podrá utilizar para que ejerza un efecto farmacológico, entre las formulaciones iniciales y las definitivas y así poder establecer la relación entre la cantidad de fármaco disponible y su velocidad de disposición con el efecto terapéutico obtenido.
Hasta finales de los años setenta, en EEUU, los fármacos genéricos habían sido comercializados sin este tipo de estudio y habían surgido bastantes problemas de seguridad y eficacia con genéricos de digoxina, fenitoína, antidepresivos tricíclicos o antidiabéticos orales. A partir de entonces la Food and Drug Administration (FDA), estableció la necesidad de la comparación farmacocinética para la demostración de la bioequivalencia entre dos formulaciones de un mismo principio activo, basada en la cantidad total de fármaco absorbida, medida como el área bajo la curva (AUC) de las concentraciones del fármaco frente al tiempo), y en la velocidad de absorción, medida como la concentración máxima alcanzada (Cmax).
Fue a principios de los años 90 cuando se establecieron los parámetros que actualmente se siguen utilizando para establecer bioequivalencia.
OBJETIVO PRINCIPAL DE LOS ESTUDIOS DE BIOEQUIVALENCIA
Los ensayos clínicos de bioequivalencia farmacocinética, a los que a partir de ahora se denominará como ensayos o estudios de bioequivalencia, tienen por objetivo demostrar que dos formulaciones de un mismo principio activo presentan un comportamiento farmacocinético tan semejante que se puede asumir, sin riesgo a equivocarse, que presentarán, de la misma forma, efectos farmacológicos igualmente semejantes, es decir, son terapéuticamente equivalentes y, por lo tanto, intercambiables. Esta afirmación se basa en el principio de que a iguales concentraciones plasmáticas de una misma sustancia corresponden iguales efectos farmacodinámicos (Figura 1).
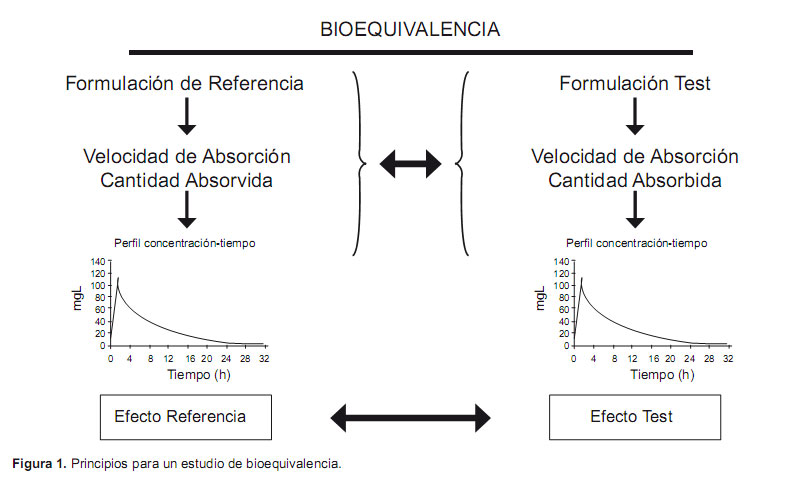
La demostración de la bioequivalencia farmacocinética es la condición necesaria, en la mayoría de los casos, para poder afirmar que dos medicamentos con la misma cantidad de un mismo principio activo producen el mismo efecto terapéutico (equivalencia terapéutica) y pueden ser responsables de la aparición de los mismos efectos adversos (seguridad); estos argumentos son imprescindibles para la autorización de la comercialización de los fármacos genéricos por parte de las autoridades sanitarias europeas y norteamericanas (5,6).
Estos dos parámetros farmacocinéticos caracterizan la biodisponibilidad de un principio activo, esto es, la velocidad y la magnitud con la que un ingrediente activo es absorbido desde un producto farmacéutico y está disponible en su lugar de acción. Su cálculo se realiza midiendo las concentraciones del fármaco en una matriz biológica fácilmente accesible, generalmente en sangre, ya que no suele ser posible medirlas en el lugar de acción. Como medida de la cantidad de fármaco absorbido se utiliza el área bajo la curva concentración-tiempo (AUC, del inglés area under the curve), y como indicador de la velocidad de absorción se mide la concentración máxima (Cmax) alcanzada en la curva concentración-tiempo y el tiempo que tarda en alcanzarse (Tmax).
Cuando dos medicamentos son equivalentes en la velocidad y cantidad del fármaco activo que se absorbe y llega al tejido o área donde se produce su efecto, se considera que son terapéuticamente equivalentes y pueden usarse indistintamente. Es decir, si se produce la equivalencia farmacocinética se asume que la misma equivalencia existirá en el plano farmacodinámico y, lo que es más importante, en la eficacia terapéutica. Así, se entiende por bioequivalencia entre dos productos cuando presentan una biodisponibilidad comparable en condiciones experimentales apropiadas. Definir a un medicamento como bioequivalente frente a otro es una cuestión de innegable relevancia desde el punto de vista de la salud pública. Por esta razón, los requisitos de la definición de bioequivalencia y las condiciones en que debe demostrarse, esto es, el tipo y características de los ensayos clínicos con los que se realizará la comparación de la biodisponibilidad, tanto en lo referente a la selección y al número de participantes en el estudio, a las dosis de los fármacos que deben ser analizadas, a la metodología del análisis farmacocinético que debe utilizarse y a las variables o parámetros farmacocinéticos que se deben evaluar y, por último, al tipo de análisis matemático al que deben someterse los resultados obtenidos en el estudio, están sujetos a normas de procedimiento bastante homogéneas tanto en el ámbito geográfico de la Unión Europea (European Medicine Agency - EMEA) como en los Estados Unidos (FDA) (3,6,7).
De acuerdo con estas normas de consenso (EMEA, FDA) (5,6) , se considera que dos formulaciones son bioequivalentes cuando la diferencia en la velocidad y la magnitud de la absorción entre ellas es inferior al 20% (diferencias medias entre formulaciones comprendidas entre 0,8 y 1,2), en términos del intervalo de confianza (IC 90%) para la proporción entre las medias de las dos formulaciones comparadas (AUC test / AUC Referencia y Cmax test / Cmax Referencia). Esto viene a significar que el 90% de la población esta incluida dentro de los límites del intervalo, que debe ser menor de 20%. Aunque en la práctica, esta diferencia suele ser mucho menor, del orden de 5% (8) lo que equivale a la variabilidad que las agencias reguladoras aceptan, como control de calidad de fabricación, entre los diferentes lotes de fabricación de un medicamento. Este límite de aceptabilidad se decidió en base a que una diferencia de 20% en las concentraciones del fármaco activo en sangre, resultado de la variabilidad permitida en las características de composición de los lotes galénicos, de circunstancias ambientales y particulares de los pacientes, no posee relevancia desde el punto de vista clínico para la inmensa mayoría de los fármacos.
En la práctica, la demostración de bioequivalencia es el método más apropiado para establecer equivalencia terapéutica entre dos fármacos que contienen excipientes que se conoce no alteran la eficacia ni la seguridad.
DISEÑO DE UN ESTUDIO DE BIOEQUIVALENCIA CLÁSICO
En un estudio de bioequivalencia, al igual que en cualquier otro ensayo clínico, una vez planteado el objetivo, se debe planificar su realización de manera que sea posible dar una respuesta adecuada, es decir cómo se debe realizar el estudio, en quién debe de efectuarse, qué dosis se debe utilizar, cuántas muestras y a qué tiempos, qué se debe medir, qué características debe tener el método de análisis de las muestras biológicas y, por último, cómo se deben analizar e interpretar los resultados obtenidos (9).
En la gran mayoría de los casos, el diseño de los estudios de bioequivalencia es el de un ensayo clínico cruzado y con asignación aleatoria de dos secuencias de tratamiento, con dos períodos y con administración de una dosis única de los fármacos en estudio en cada uno de los periodos (diseño cruzado 2x2) (Figura 2).
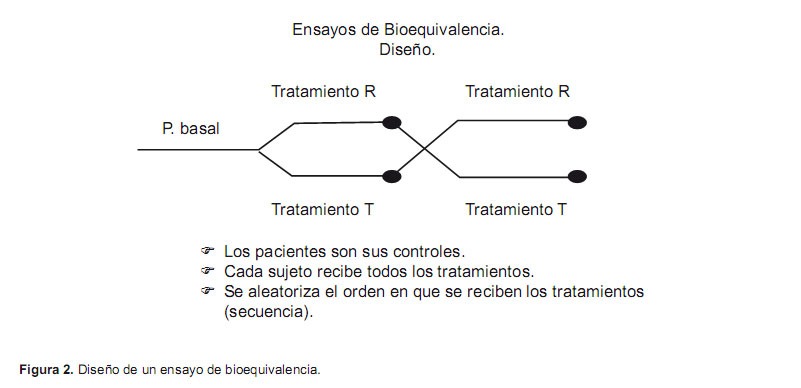
Los participantes en el ensayo reciben las dos formulaciones que se estudian, pero en diferente orden. Lo que se asigna aleatoriamente no es el fármaco, sino el orden en el que lo recibirán, es decir la secuencia de tratamiento (secuencia referencia-test o secuencia testreferencia). Cada día del estudio se denomina periodo, y en él, se administra una dosis única de cada una de las formulaciones, generalmente en ayunas. Entre cada administración de fármaco existe un periodo de lavado de una duración suficiente para permitir que se hayan eliminado del organismo todo el medicamento y sus metabolitos antes de administrar la segunda dosis. Habitualmente, es suficiente esperar un mínimo de cinco vidas medias para asegurar la completa eliminación y evitar efectos residuales de las formulaciones. En la mayoría de los fármacos, la variabilidad intrasujeto es menor que la intersujeto, por lo que el diseño cruzado permite un tamaño muestral menor, ya que cada sujeto actúa como control de sí mismo, eliminando la variabilidad interindividual (5).
Todas las condiciones ambientales que rodean la realización del estudio y a sus participantes se estandarizan al máximo posible para reducir fuentes de variabilidad no controlada. Así, la ingesta de líquidos y la dieta son iguales todos los días del estudio, el ejercicio físico se reduce a los mínimos imprescindibles, se prohíbe la ingesta de alimentos o bebidas que pudieran modificar el comportamiento farmacocinético, como bebidas alcohólicas o productos que contengan xantinas y el consumo de drogas de abuso, porque pueden modificar e interferir con los procesos metabólicos de los fármacos y por tanto modificar su biodisponibilidad y por último, la administración de los fármacos en estudio de realiza de forma estandarizada e idéntica a todos los participantes. Habitualmente, los ensayos de bioequivalencia se realizan en condiciones de ayunas de al menos diez horas.
SELECCIÓN DE LOS PARTICIPANTES
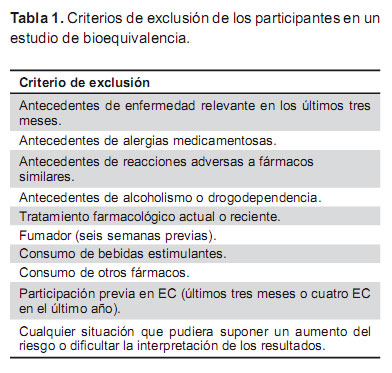
Los criterios que se aplican a la selección de los participantes en los estudios de bioequivalencia tienen como objetivo reducir la variabilidad aportada por las características demográficas y antropométricas de los participantes o por situaciones patológicas, de manera que si aparecen diferencias relevantes en el comportamiento farmacocinético de los medicamentos estudiados éstas no puedan ser atribuidas a la heterogeneidad de los participantes, sino a que realmente los fármacos se comportan de manera diferente. Por este motivo se eligen voluntarios sanos, que pueden ser de ambos sexos, de peso normal o de índice de masa corporal dentro de límites normales, de edades comprendidas entre 18 y 55 años, no fumadores ni bebedores (Tabla 1) (6,10).
CÁLCULO DEL TAMAÑO MUESTRAL
El número de voluntarios que deben participar se calcula fundamentalmente a partir de la variabilidad interindividual descrita para los parámetros principales de evaluación de la biodisponibilidad (AUC y Cmax), que se puede obtener a partir de estudios piloto, de ensayos clínicos previos o de datos disponibles en la literatura científica (11,12).
Por regla general, cuanto mayor es la variabilidad de los parámetros farmacocinéticos es necesario un mayor número de participantes. El tamaño muestral de los ensayos de bioequivalencia es el principal factor del que depende la probabilidad de concluir erróneamente que dos formulaciones no son bioequivalentes. Según la EMEA, el número de sujetos, nunca debería ser inferior a 12, aunque lo habitual es sobrepasar este número hasta un tamaño de 24 o 36 participantes.
SELECCIÓN DE LA DOSIS POR ADMINISTRAR
La selección de las dosis por utilizar en estos ensayos clínicos, viene determinada, al menos, por tres aspectos relevantes, que en la mayoría de las situaciones se valoran simultáneamente:
-
La dosis seleccionada ¿es suficientemente segura?
-
Con la dosis que se ha administrado, ¿tendrá el método analítico que se utilice para la cuantificación de los fármacos la suficiente precisión?
-
La dosis seleccionada, ¿será la más sensible para la detección de diferencias relevantes en las características farmacocinéticas?
Deben seleccionarse siempre dosis comprendidas dentro del rango de las dosis del fármaco de referencia autorizadas para su comercialización. En ocasiones, la selección de cuál de las dosis se debe estudiar, está más condicionada por la importancia relativa de alguna o varias de las circunstancias mencionadas anteriormente. Por ejemplo, si un fármaco de referencia presenta una relación lineal entre las diferentes dosis autorizadas y las variables farmacocinéticas principales, el criterio de selección de la dosis sólo tendrá como limitación la tolerabilidad del fármaco y las características del método de análisis químico por este orden, ya que todas las dosis que se podrían utilizar tendrían un comportamiento farmacocinético semejante (5,7,9)
ANÁLISIS FARMACOCINÉTICO
Después de la administración de cada formulación es necesario saber qué cantidad del fármaco existe en el organismo y cómo va cambiando a lo largo del tiempo. El procedimiento más habitual consiste en la obtención de sucesivas muestras de sangre mediante sistemas adecuados para reducir el número de punciones venosas en los participantes. Es mucho menos frecuente que sea necesaria la determinación del fármaco en otras matrices o fluidos biológicos, como por ejemplo la cuantificación y comparación del perfil de excreción urinaria; cuando esto ocurre, se debe casi sin excepción a que la cuantificación de los principios activos en sangre total, plasma o suero, no permite una buena definición de los parámetros farmacocinéticos, ya sea porque el fármaco no alcance concentraciones suficientes para su adecuada medición, o porque su paso por sangre sea demasiado fugaz como para poder realizar una definición adecuada de su relación concentración/tiempo.
El número de muestras y los tiempos en los que se obtienen deben ser los adecuados para definir el perfil de la curva concentración-tiempo y sus distintas fases (absorción, distribución, metabolismo y eliminación) de forma que se pueda caracterizar adecuadamente la Cmax y el momento en que aparece (Tmax) y al menos el 80% del AUC total. Habitualmente, es suficiente con la obtención de entre 12 y 18 muestras para cada formulación, que se deberían prolongar durante al menos tres vidas medias, aunque lo deseable es que se prolongue hasta cinco vidas medias (9,10) .
Una vez extraídas, las muestras se deben procesar y conservar adecuadamente y con arreglo a las instrucciones del laboratorio analítico, que garanticen la conservación óptima de la sustancia a cuantificar en la matriz biológica en la que está contenida.
El método analítico debe estar perfectamente validado; es imprescindible que reúna las condiciones de precisión (sensibilidad y especificidad) y reproducibilidad adecuadas para garantizar que los resultados que se obtienen corresponden realmente a lo que se desea medir. Como es lógico, lo más frecuente es medir la concentración plasmática del fármaco administrado. En las ocasiones en que la cuantificación del fármaco no es posible, ya sea porque sus concentraciones son muy bajas o porque la vida media es muy corta, puede justificarse la medida de la concentración de aquellas sustancias directamente derivadas del fármaco (metabolitos) que reflejen adecuadamente la biodisponibilidad de la sustancia activa. El sentido general de las normas de consenso sobre bioequivalencia da preferencia a la cuantificación del fármaco original (sin biotransformar) siempre y cuando sea cuantificable, ya que su perfil farmacocinético refleja mejor el comportamiento de las formulaciones y ofrece una mayor capacidad de detectar diferencias entre ellas, (p.e. la Cmax de un metabolito es mucho mas variable que la del fármaco del que deriva) independientemente de que sea una molécula activa o no. Esto resulta especialmente cierto cuando los metabolitos se forman mediante procesos químicos saturables, en los que la cuantificación y comparación de los metabolitos es mucho menos sensible que la de la molécula original (3,7,10).
Los parámetros farmacocinéticos sobre los que se basará la afirmación de bioequivalencia, se calculan a partir de la curva de las concentraciones del fármaco durante el tiempo en el que se extraen las muestras, a partir de la administración de cada una de las formulaciones. Mediante un análisis independiente de modelo en el que se realiza un ajuste lineal para el cálculo de la constante de eliminación y por tanto de la vida media de eliminación y del AUC entre 0 e infinito (Figura 3).
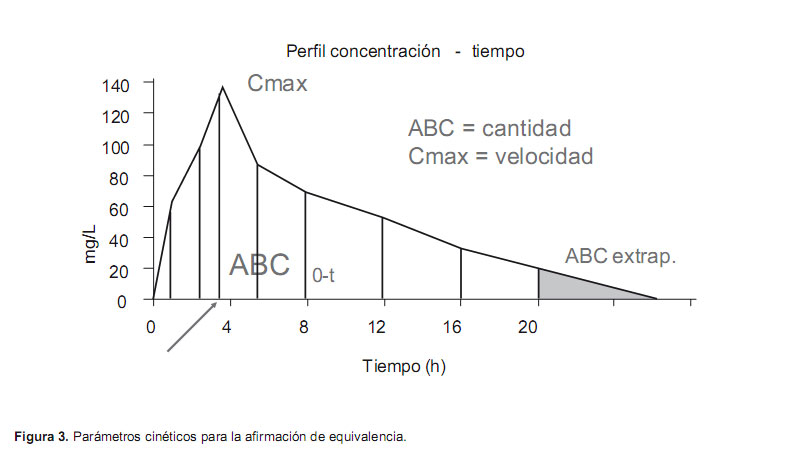
Según las recomendaciones de las agencias reguladoras (EMEA, FDA) (6,7), los parámetros farmacocinéticos adecuados para el estudio de la bioequivalencia son:
-
El área bajo la curva concentración-tiempo (AUC) calculada por el método trapezoidal, que se puede medir desde la administración del fármaco hasta la última muestra con concentración cuantificables (AUC0-t) o extrapolándola hasta que la concentración llegue a cero (AUC0-∞). Esta extrapolación se calcula como prolongación de la recta de la fase de eliminación (última concentración medida dividida por la constante de eliminación) y no debe ser superior al 20% del área bajo la curva que describen los valores de las concentraciones del fármaco desde el momento de su administración hasta el tiempo en el que aparece la última concentración cuantificable. El AUC refleja la cantidad de fármaco biodisponible.
-
La concentración máxima (Cmax) y el tiempo en el que se alcanza (Tmax), obtenidas directamente de las concentraciones plasmáticas y que reflejan la velocidad con la que el fármaco puede ser utilizado por el organismo.
La vida media de eliminación es útil para comparar los perfiles cinéticos entre las formulaciones, comprobar la existencia de concordancia con lo descrito en la literatura y valorar si el periodo de lavado ha sido suficiente.
El AUC y la Cmax son los parámetros primarios para evaluar bioequivalencia, y la Tmax y la vida media constituyen los parámetros secundarios.
ANÁLISIS ESTADÍSTICO
La principal preocupación de las autoridades sanitarias es el riesgo que supondría para los pacientes la aceptación errónea de que un producto sea bioequivalente cuando en realidad no lo es. Por este motivo solamente se deben utilizar procedimientos estadísticos en los que este riesgo no exceda del 5% aceptable. El riesgo de que no se pueda concluir que dos formulaciones son bioequivalentes cuando en realidad lo son, es menos preocupante desde el punto de vista de la salud pública, ya que en este caso el fármaco no estaría autorizado para su dispensación y, por ello, se acepta que pueda existir una probabilidad de hasta el 20% en que el fármaco no pueda demostrar que es bioequivalente aun siéndolo. El objetivo de los estudios de bioequivalencia no consiste en la demostración de superioridad de una formulación frente a la otra, sino de que ambas sean prácticamente indistinguibles y en este caso la hipótesis nula es que las dos formulaciones son diferentes; así pues, si asumimos un error alfa de 0,05 la probabilidad de que se concluya erróneamente que dos formulaciones son bioequivalentes si no lo son, es de sólo 5% (8). Con un poder de 80% tendremos una probabilidad de 20% de no ser capaces de demostrar la bioequivalencia de dos formulaciones que realmente sí lo son.
Cuando realizamos un estudio de bioequivalencia, las diferencias encontradas entre las formulaciones pueden ser debidas a diferentes factores:
-
La secuencia u orden de administración de las formulaciones (efecto secuencia);
-
El período de administración (efecto período);
-
La formulación que se ha administrado (efecto formulación);
-
El posible efecto del fármaco administrado en el primer periodo sobre el fármaco administrado en el segundo periodo (efecto de arrastre o carry-over).
-
El diferente comportamiento del fármaco en los individuos a los que se administra (variabilidad interindividual);
-
Por último, otro factor que, a diferencia de los anteriores, no esta controlado mediante los requisitos o peculiaridades del diseño del estudio y que corresponde a un conjunto de factores aleatorios o variabilidad residual (intraindividual).
Se está, por tanto, ante los resultados de un estudio experimental en los que de forma simultánea intervienen múltiples factores; unos, los más importantes, controlados en el diseño y características del estudio y otros (la variabilidad intraindividual o residual) de carácter aleatorio. Lógicamente, el análisis de los resultados debe realizarse mediante los métodos matemáticos que permitan identificar en qué magnitud contribuyen todos estos factores al resultado final del estudio y si existe algún o algunos de ellos que influyan de forma decisiva (estadísticamente significativa) en la obtención de una conclusión afirmativa o negativa sobre la bioequivalencia de las formulaciones, por lo que será obligada la utilización de técnicas estadísticas multivariadas, a partir de modelos generales lineales (GLM), que en este caso será un análisis de varianza multivariante (MANOVA). De esta forma se puede comprobar que no existe efecto periodo, secuencia, de arrastre ni de formulación.
La comprobación de que alguno de estos factores, a excepción de la variabilidad interindividual, tienen un efecto significativo desde el punto de vista matemático, sobre los resultados del análisis multivariado, indica de forma prácticamente constante, que existe algún problema en la realización del estudio: diferencias en la obtención, manejo, almacenamiento y análisis de las muestras biológicas, diferencias climáticas, dietéticas o de actividad física, entre otras. La trascendencia de la significación de estos efectos puede ser de tal magnitud que obligue a la realización de análisis de sensibilidad de los resultados del estudio, mediante la utilización de técnicas estadísticas no paramétricas o a que los resultados del estudio no puedan considerarse aceptables y por tanto no sean válidos, ya que implican un error grave en el diseño (5) .
Sin embargo, aunque este método resulta suficientemente robusto y discriminativo, solamente informa de si existen o no diferencias matemáticamente relevantes, y no de la magnitud de esas diferencias, que será el elemento clave para poder evaluar si los resultados del estudio permiten concluir que los fármacos estudiados son o no bioequivalentes. Para que dos productos puedan ser considerados como bioequivalentes no sólo se requiere, por tanto, que no existan diferencias estadísticamente significativas entre sus parámetros farmacocinéticos, sino que además la magnitud de estas diferencias no exceda los límites que marca el intervalo de confianza de aceptabilidad de estas diferencias, por lo que se requiere que el intervalo de confianza del 90% para la diferencia entre las medias de las dos formulaciones (AUC y Cmax) no sea ni superior ni inferior a ±20% para el cociente entre los valores medios de los parámetros farmacocinéticos de las dos formulaciones, esto es, que esté comprendido entre 80 y 120%. Dado que los valores de AUC y Cmax no suelen seguir una distribución normal, se recomienda la transformación logarítmica de estos parámetros. Para los datos con transformación logarítmica, los límites del intervalo de confianza del 90% se sitúan en 80-125 para poder concluir bioequivalencia.
La evaluación estadística de la Tmax sólo tiene sentido como criterio principal de bioequivalencia cuando una velocidad de liberación del principio activo más o menos rápida puede relacionarse con un efecto clínico relevante, o con la aparición de efectos adversos. Como la Tmax es una variable discontinua que depende de los puntos de extracción prefijados, no es adecuado realizar un análisis paramétrico como el ANOVA, sino que se suele calcular el intervalo de confianza por métodos no paramétricos.
Si se tiene en cuenta que es la amplitud del intervalo de confianza el criterio fundamental para la confirmación de la hipótesis de bioequivalencia es difícil que existan diferencias grandes entre dos genéricos.
CONCLUSIONES
La consideración como genérico de un medicamento y, por tanto, como intercambiable con su fármaco de referencia descansa sobre los resultados obtenidos al comparar de manera rigurosa y con arreglo a una detallada serie de normas internacionales de carácter técnico, su comportamiento farmacocinético mediante ensayos clínicos de bioequivalencia.
Estas normas definen con suficiente precisión la forma en que deben realizarse estos ensayos, su diseño, cuáles deben ser los parámetros de evaluación y cómo debe realizarse el análisis de los resultados obtenidos, así como los límites de aceptación para confirmar o rechazar que las formulaciones estudiadas puedan considerarse como equivalentes.
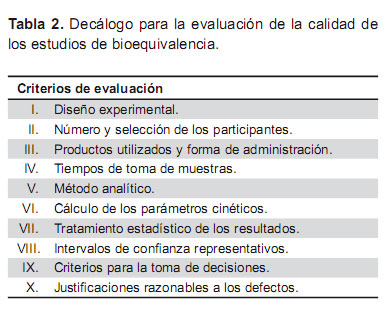
El objetivo final de todo este proceso tiene como único sentido poner a disposición de la sociedad, fármacos de calidad que, además, puedan contribuir a un uso más racional de los recursos económicos en el sistema sanitario (Tabla 2).
Conflictos de interés
Los autores declaran no tener conflictos de interés para la publicación de este artículo.
REFERENCIAS BIBLIOGRÁFICAS
1. España, Ministerio de Sanidad y Consumo. Plan estratégico de política farmacéutica para el sistema nacional de salud español. Madrid: Ministerio de Sanidad y Consumo; 2004.
2. España, Ministerio de Sanidad y Consumo. Garantías y uso racional de medicamentos y productos sanitarios, Ley 29/2006. Madrid: Ministerio de Sanidad y Consumo; 2006.
3. European Medicines Agency, Committee, Committee for Medicinal Products for Human Use. Guideline on the investigation of bioequivalence(Draft). London: EMEA; 2008. CPMP/ EWP/QWP/1401/98 Rev.1
4. Diez-Rodríguez MV. Genéricos. Claves para su conocimiento y comprensión. Madrid: Editores Médicos; 1999.
5. Guerra P, Lubomirov R. Medicamentos genéricos y estudios de bioequivalencia. En: Govantes J, Fernández P, Govantes C, editores. Manual Normon. 8ª Ed. Madrid: Laboratorios Normon; 2007.
6. Center for Drug Evaluation and Research. Guidance for industry: bioavailability and bioequivalence studies for orally administered drug products-general considerations. Maryland: Food and Drug Administration; 2000.
7. European Medicines Agency, Committee for Proprietary Medicinal Products. Note for guidance on the investigation of bioavailability and bioequivalence. London: EMEA; 2001. CPMP/EWP/ QWP/1401/98.
8. Ramirez E, Guerra P, Laosa O, Duque B, Tabares B, Lei SH, et al. The importance of sample size, log-mean ratios, and intrasubject variability in the acceptance criteria of 108 bioequivalence studies. Eur J Clin Pharmacol 2008; 64 (8): 783-93.
9. Abad F, Martínez E, Gálvez MA. Estudios de bioequivalencia: análisis y aspectos metodológicos. En: García AG, Gandía L. El ensayo clínico en España. Madrid: Farmaindustria; 2001.
10. European Medicines Agency, Committee for Proprietary Medicinal Products. Questions & answers on the bioavailability and bioequivalence guideline. London: EMEA; 2006. EMEA/CHMP/ EWP/40326/2006.
11. Julious SA. Tutorial in biostatistics: sample sizes for clinical trials with normal data. Statistic Med. 2004; 23(12): 1921-86.
12. Wegener HH, Vögtle-Junkert U. Intrasubject variability in bioequivalence studies ilustrated by the example of ibuprofen. Int J Clin Pharmacol Ther. 1996; 34(1): 21-31.
Correspondencia: Jesús Frías Iniesta. Centro de Farmacología Clínica, Hospital Universitario La Paz, Facultad de Medicina, Universidad Autónoma de Madrid
Dirección: Paseo de la Castellana, 261 28046, Madrid, España
Correo electrónico: jesus.frias@uam.es
Recibido: 03-11-09
Aprobado: 16-12-09

