










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
Print version ISSN 1726-4634
Rev. perú. med. exp. salud publica vol.29 no.4 Lima Oct./Dec. 2012
Sección Especial
Eficacia y seguridad en la nueva regulación de productos farmacéuticos en el Perú
Efficacy and safety in the new regulation of pharmaceutical products in Peru
Hans Vásquez1,a, Sofía Salas1,b, Linder Figueroa1,2,c, Sonia Gutiérrez1,b
1 Dirección General de Medicamentos, Insumos y Drogas (DIGEMID), Ministerio de Salud. Lima, Perú.
2 Unidad de Posgrado de Farmacia y Bioquímica, Universidad Nacional Mayor de San Marcos. Lima, Perú.
a Médico cirujano especialista en Salud Pública con mención en Epidemiología; b químico farmacéutico; c químico farmacéutico magister en Farmacia Clínica
RESUMEN
A nivel internacional los productos farmacéuticos son autorizados luego de evaluar el balance riesgo-beneficio, teniendo en cuenta parámetros de eficacia, seguridad y calidad. A partir del 2009, en el Perú se ha establecido que todo PF por comercializar deberá contar con evidencia de eficacia y seguridad. Según la nueva reglamentación de la ley, vigente a partir del 2012, las especialidades farmacéuticas (EF) se han agrupado en tres categorías: categoría 1 si se encuentran en el Petitorio Nacional de Medicamentos Esenciales, categoría 2 si están autorizados en países de alta vigilancia sanitaria y categoría 3 si no se incluyen en las categorías 1 o 2. La documentación científica a presentar al momento de su inscripción o reinscripción en la entidad reguladora dependerá de la categoría de la EF.
Palabras clave: Evaluación de medicamentos; Legislación de medicamentos; Medicamentos esenciales (fuente: DeCS BIREME).
ABSTRACT
Internationally, pharmaceutical products (FP) are authorized after their risk-benefit profile has been assessed, taking into account efficacy, safety and quality parameters. In 2009, it was established in Peru that all pharmaceutical products to be commercialized should show proof of their efficacy and safety. According to the new regulation, in effect as of 2012, the pharmaceutical specialties (FS) have been grouped into three categories: category 1, if included in the National List of Essential Medicines; category 2, if authorized in countries with high health surveillance; and category 3, if not included in categories 1 or 2. The scientific documentation to be submitted for the registration or re-registration of the product in the regulatory entity will depend on its FS category.
Key words: Drug evaluation; Legislation, drug; Drugs, essential (source: MeSH NLM).
INTRODUCCIÓN
A nivel mundial son las autoridades reguladoras de medicamentos (ARM) las que autorizan el uso y comercialización de los productos farmacéuticos (PF) después de haber realizado una evaluación del balance riesgo-beneficio (1), basada en estándares de calidad, eficacia y seguridad (2,3). Estas dos últimas se evalúan a partir de datos derivados de estudios preclínicos y clínicos; y para garantizar que se mantenga un balance positivo se toman medidas para minimizar los posibles riesgos durante el uso de los medicamentos. Aun así, es necesario realizar una vigilancia posterior al inicio de la comercialización, y en algunos casos diseñar estudios que proporcionen información adicional sobre el perfil de seguridad del producto a largo plazo y su real efectividad en la práctica clínica, ya que los ensayos clínicos suelen realizarse en condiciones ideales y especificas (4,5).
Durante el proceso de evaluación de un PF es importante valorar toda la evidencia científica disponible, incluyendo aquellos estudios que no favorecieron al producto evaluado (6). Es válido utilizar información de otras instituciones científicas o ARM de referencia a nivel mundial (7,8). Los aspectos de la calidad de un PF (que incluye el producto terminado, el principio activo y los excipientes) relacionados a aspectos farmacéuticos, químicos, de fabricación y controles de calidad también son fundamentales e influyen en los aspectos de eficacia y seguridad (9). En el Perú se denomina PF a cualquier producto que sea un medicamento, producto biológico, medicamento herbario, producto dietético o edulcorante, y los productos galénicos.
LA NUEVA REGULACIÓN
Antes de la nueva regulación, el tiempo que tenía la ARM (Dirección General de Medicamentos, Insumos y Drogas - DIGEMID) para autorizar un medicamento era solamente de siete días, y todos los PF (incluyendo los productos biológicos) eran evaluados para su autorización en base a requisitos mínimos tales como algunos datos de control de calidad y ninguna evidencia científica de eficacia o seguridad; aspectos no acordes con prácticas y estándares recomendados a nivel internacional.
La situación antes mencionada generó la necesidad de actualizar y mejorar la regulación, entonces se establecieron nuevos requisitos para la autorización de un PF y la vigilancia posterior a la comercialización. Esta experiencia no ha sido ajena a otros países que también han tenido que introducir nuevas regulaciones en algún momento de su historia (10-12).
Legalmente, la nueva regulación empezó a gestarse con la Ley 29316 aprobada en enero de 2009, en la cual se modificó el artículo 50 de la Ley General de Salud (la que establecía requisitos mínimos para autorizar un medicamento). Esta ley surgida como consecuencia del Acuerdo de Promoción Comercial Perú-Estados Unidos (también llamado Tratado de Libre Comercio), estableció por primera vez requisitos de eficacia y seguridad para la autorización de medicamentos.
En noviembre de 2009 se publicó la Ley 29459 Ley de Productos Farmacéuticos, Dispositivos Médicos y Productos Sanitarios (13). En ella se establecen fundamentos y criterios técnicos más específicos para evaluar los productos farmacéuticos, en base a evidencia de eficacia, seguridad y calidad. Esta Ley no incluye a todos los PF, como por ejemplo los productos biológicos, los cuales se desarrollan en su reglamento correspondiente. En julio de 2011, se publicó el reglamento de la Ley 29459 (DS 016-2011-SA) (14).
REQUISITOS DE EFICACIA Y SEGURIDAD DE MEDICAMENTOS
Dentro de las nuevas regulaciones sobre requisitos de eficacia y seguridad se ha establecido una distinción entre información técnica, estudios y sustento. Cuando se usa el término estudios, se refiere a datos clínicos (además de preclínicos) que deben ser presentados como evidencia de eficacia y seguridad, el término información se refiere a otro tipo de evidencia científica (no necesariamente datos clínicos de estudios) y cuando se usa el término sustento la regulación se refiere a cualquier tipo de evidencia que puede incluir estudios u otro tipo de evidencia científica.
Todos los aspectos anteriormente señalados tienen como fundamento, principios (señalados en la Ley) que sirven de criterio interpretativo para el proceso de evaluación o revisión realizada por la ARM (DIGEMID). Se tiene el principio de eficacia, que es el beneficio de la persona basado en estudios clínicos y preclínicos; el principio de seguridad, que significa que el producto es usado en condiciones reales sin presentar riesgo para la salud; el principio de objetividad, que significa que las acciones y decisiones son sustentadas en información científica independiente y objetiva. Otros principios considerados son: calidad, accesibilidad, racionalidad, equidad, bien social y transparencia.
Las especialidades farmacéuticas (EF) incluyen a la mayor cantidad de medicamentos autorizados y usados en la práctica clínica, no estando en este grupo los agentes de diagnóstico, radiofármacos y gases medicinales. Las EF se dividen en tres categorías según el principio activo, también llamado ingrediente farmacéutico activo (IFA). La categoría no va necesariamente a la par con la necesidad de una menor o mayor evidencia en el proceso de inscripción o reinscripción (Tabla 1).
Las EF categoría 1 son aquellos productos cuyo IFA se encuentran en productos del Petitorio Nacional de Medicamentos Esenciales (PNME). Los productos de esta categoría que son iguales (en forma farmacéutica, concentración y vía de administración) a uno del PNME, no tienen como requisito presentar estudios o información de eficacia o seguridad. Si es de categoría 1, pero el producto no es igual a uno del PNME (está en otra forma farmacéutica, otra concentración u otra vía de administración) tendrá que sustentar eficacia y seguridad (siendo de categoría 1).
Por ejemplo: el paracetamol de 500 mg tableta vía oral es un producto que está en el PNME. Una empresa podría solicitar el registro sanitario (caso hipotético) de paracetamol en concentración de 2 g para vía endovenosa o intratecal. Este último producto sería categoría 1 por tener al paracetamol como mismo principio activo, y deberá sustentar eficacia y seguridad por presentar diferente concentración, forma farmacéutica y vía de administración; es decir, solamente en caso el producto sea igual (en todas sus características) al del PNME no tiene como requisito presentar información, estudios o sustento.
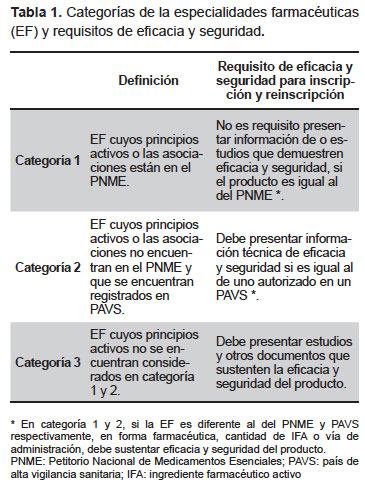
Las EF categoría 2, son aquellas cuyo IFA es el mismo que el de un producto aprobado en un país de alta vigilancia sanitaria (PAVS): Estados Unidos de América, Reino Unido, Francia, Canadá, Japón, Australia, España, Italia, Alemania, Bélgica, Holanda, Noruega, Suiza, Suecia y Dinamarca. Estos productos tienen como requisito presentar información técnica de eficacia y seguridad; pero si este mismo IFA está en otra forma farmacéutica u otra concentración u otra vía de administración, tendrá que sustentar eficacia y seguridad del producto; situación similar a los de categoría 1.
Por ejemplo: un producto de forma farmacéutica de tableta oral que contiene isoniazida 50 mg, más rifampicina 120 mg, más pirazinamida 300 mg, está autorizado en Estados Unidos, Reino Unido, entre otros PAVS. Sería un producto categoría 2 porque los mismos principios activos están contenidos en un producto autorizado en un PAVS y tiene como requisito presentar la información de eficacia y seguridad.
Si una empresa solicita el registro sanitario (caso hipotético) de un producto en forma de tableta con los mismos principios activos, pero ha cambiado la cantidad de IFA de la isoniazida a 100 mg y el resto sigue igual, este producto seguiría siendo categoría 2 (a pesar que como producto no es el mismo que el de un PAVS) ya que los principios activos están incluidos en un producto de un PAVS pero debe sustentar eficacia y seguridad.
Las EF categoría 3, que incluye a los productos cuyos IFA no están en el PNME o no han sido aprobados en PAVS, deben presentar estudios preclínicos y clínicos que sustenten la eficacia y seguridad del producto. Existen estándares internacionales para presentar la documentación de estudios preclínicos y clínicos (15-17).
Cualquier producto que contenga un IFA o una combinación de dos o más IFA, que no estén contenidos en un producto del PNME o de un PAVS, será de categoría 3. Aun cuando cada uno de los IFA (de una combinación) pueden estar en un producto que está en el PNME o PAVS como monofármaco, si no están ambos a la vez en un producto que está en el PNME o PAVS, correspondería a categoría 3.
En caso de reinscripción de productos de categoría 3 se solicitará información técnica de eficacia y seguridad, a aquellos productos que a la fecha de entrada en vigencia de la Ley 29459 cuenten con registro sanitario vigente. En las sucesivas reinscripciones no será necesario presentar adicional información, salvo que haya alguna modificación en el proceso de fabricación o se proponga alguna modificación en la ficha técnica, los cuales deberán ser sustentados con evidencia científica, si es necesario.
El plazo vigente para evaluar medicamentos categoría 1 es de 60 días calendario; 90 días calendario para medicamentos categoría 2, y 12 meses para los de categoría 3.
ESTUDIOS DE EQUIVALENCIA TERAPÉUTICA
Todas las EF deben presentar estudios que demuestren equivalencia terapéutica, según se establezca en la directiva específica. Este requisito será necesario para los productos genéricos o multifuente.
Dentro de los requisitos están los estudios de equivalencia terapéutica para demostrar la intercambiabilidad. Según la Organización Mundial de la Salud (OMS), un producto intercambiable es aquel que es equivalente terapéutico a un producto comparador y puede ser intercambiado con el comparador en la práctica clínica (18).
La manera de demostrar equivalencia terapéutica (en caso se requiera) es a través de estudios de bioequivalencia, farmacodinamia, estudios clínicos o estudios in vitro.
APUNTES FINALES
La nueva regulación constituye un avance y un reto para la Autoridad Nacional de Medicamentos, la industria farmacéutica, los profesionales de salud y público en general (19). El sustento de gran parte de estas nuevas regulaciones, son criterios y requisitos recomendados por la OMS y recoge la experiencia internacional.
Las nuevas regulaciones necesitan ser implementadas de manera progresiva, y estar abiertos y en constante diálogo con los avances científicos y regulatorios a nivel internacional, teniendo en cuenta la realidad local, pero siempre teniendo como horizonte principal que la población disponga de productos farmacéuticos de calidad, efectivos y seguros.
Agradecimientos: al Dr. Germán Málaga por la revisión del artículo, sus valiosos aportes y sugerencias.
Descargo de responsabilidad: las opiniones y afirmaciones contenidas en el presente artículo son propias de los autores y no necesariamente representan la opinión de la Dirección de Medicamentos, Insumos y Drogas (DIGEMID) del Ministerio de Salud de Perú.
Referencias Bibliográficas
1. European Union, European Medicines Agency. Benefit-risk methodology project [Internet]. London: EMA; 2010 [citado el 16 de mayo 2012]. Disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/Re-port/2010/10/WC500097750.pdf [ Links ]
2. 14th International Conference of Drug Regulatory Authorities. Improving drug regulation as part of health systems stren-gthening [Internet]. Health Sciences Authority of Singapore, World Health Organization. Singapore, 30 November to 3 December 2010 [citado el 16 de mayo del 2012]. Disponible en: http://www.who.int/medicines/areas/quality_safety/regulation_legislation/icdra/14-ICDRA_recommendations.pdf [ Links ]
3. World Health Organization. Assessing national medicines regulatory systems [Internet]. Geneva: WHO; c2012 [citado el 16 de mayo del 2010]. Disponible en: http://www.who.int/medicines/areas/quality_safety/regulation_legisla-tion/assesment/en/index.html 4. Staffa JA, Dal Pan GJ. Regulatory innovation in postmarketing risk assessment and management. Clin Pharmacol Ther. 2012;91(3):555-7. [ Links ] [ Links ]
5. World Health Organization. The importance of pharmacovigilance. Geneva: WHO; 2002. [ Links ]
6. Australian Government Department of Health and Ageing, Therapeutic Goods Administration. Guidelines for level and kinds of evidence to support indications and claims. Woden: TGA; 2011. [ Links ]
7. US Food and Drug Administration. Guidance for Industry. Providing clinical evidence of effectiveness for human drug and biological products. Silver Spring: FDA; 1998. [ Links ]
8. World Health Organization. WHO Expert Committee on Specifications for Pharmaceuticals Preparations. Guidelines for registration of fixed dose combination medicinal products (Annex 5 to TRS 929). Geneva: World Health Organization; 2005. [ Links ]
9. International Conference on Harmonisation. The Common Technical Document. Efficacy-M4E(R1) [Internet]. Washington, DC: ICH; 2002 [citado el 16 de mayo del 2012]. Disponible en: http://www.ich.org/fileadmin/Pu-blic_Web_Site/ICH_Products/CTD/M4__R1__Efficacy/M4E__R1_.pdf [ Links ]
10. US Food and Drug Administration. Guidance for FDA Staff and Industry. Marketed Unapproved Drugs. Compliance Policy Guide. Silver Spring: FDA; 2011. [ Links ]
11. Rägo L, Santoso B. Drug regulation: history, present and future. En: van Boxtel CJ, Santoso B, Edwards IR. Drug benefits and risks: international textbook of clinical pharmacology. Amsterdam: IOS Press and Uppsalla Monitory Centre; 2008. [ Links ]
12. Avorn J. Two centuries of assessing drug risks. N Engl J Med. 2012;367(3):193-7. [ Links ]
13. Perú, Congreso de la República. Ley 29459. Ley de Productos Farmacéuticos, Dispositivos Médicos y Productos Sanitarios. Lima: Congreso de la República; 2009. [ Links ]
14. Perú, Ministerio de Salud. Decreto supremo 016 – 2011 - SA. Aprueban Reglamento para el Registro, Control y Vigilancia Sanitaria de Productos Farmacéuticos, Dispositivos Médicos y Productos Sanitarios. Lima: MINSA; 2011. [ Links ]
15. International Conference of Harmonisation. The Common Technical Document [Internet]. Washington, DC: ICH; c2012 [citado el 16 de mayo del 2012].. Disponible en: http://www.ich.org/products/ctd.html [ Links ]
16. International Conference of Harmonisation. Structure and content of clinical study report [Internet]. Washington, DC: ICH; c2012 [citado el 16 de mayo del 2012]. Disponible en: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E3/E3_Guideline.pdf [ Links ]
17. International Conference of Harmonisation. General Overview of eCTD [Internet]. Washington, DC: ICH; 2011 [citado el 16 de mayo del 2012]. Disponible en http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidis-ciplinary/M8/ICH_M8_eCTD_14_June_2011.pdf [ Links ]
18. World Health Organization. WHO Expert Committee On Specifications for Pharmaceutical Preparations, Fortieth Report. (WHO Technical Report Series, Nº 937) Geneva, World Health Organization, 2006. Annex 7, Multisource (Generic) Pharmaceuticals Products: Guidelines On Registration Requirements to Establish Interchangeability. [ Links ]
19. Vasquez H, Salas S, Figueroa L, Gutierrez S. Retos para las nuevas regulaciones de productos farmacéuticos en el Perú [carta]. Rev Peru Med Exp Salud Publica. 2011;28(3):567-9. [ Links ]
Correspondencia: Hans Vásquez Soplopuco
Dirección: calle Odriozola 103. Lima 27, Perú.
Teléfono: (51) 995943277
Correo electrónico: hvasquezhans@yahoo.com
Recibido: 23-08-12
Aprobado: 14-11-12
