










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
versión impresa ISSN 1726-4634
Rev. perú. med. exp. salud publica vol.30 no.2 Lima abr. 2013
Reporte de caso
Enfermedad de Kennedy en el Perú: Primeros casos con diagnóstico molecular
Kennedy disease in Peru: First cases with molecular diagnosis
Víctor Gómez-Calero 1,2,a, Mario Cornejo-Olivas 2,3,b, Olimpio Ortega 2,c, Victoria Marca 2,d, Saúl Lindo-Samanamud 2,d, Martha Flores 1,b, Luis Torres-Ramírez 1,b, Pilar Mazzetti 2,b
1 Departamento de Enfermedades Neurodegenerativas, Instituto Nacional de Ciencias Neurológicas. Lima, Perú.
2 Centro de Investigación en Neurogenética, Instituto Nacional de Ciencias Neurológicas. Lima, Perú.
3 Northern Pacific Global Health Research Fellows Training Consortium. Bethesda, EE. UU.
a Médico residente de Neurología; b médico neurólogo; c biólogo genetista; d magíster en Bioquímica
Resumen
La enfermedad de Kennedy es un trastorno neurodegenerativo de herencia recesiva ligada al cromosoma X, de inicio en la adultez, caracterizado por degeneración progresiva de las neuronas motoras espinales, debido a una mutación dinámica del gen del receptor de andrógeno. Se presentan tres familias (cinco casos) con temblor, calambres, debilidad muscular generalizada lentamente progresiva con atrofia, afectación de músculos bulbares y alteraciones endocrinas. El estudio neurofisiológico demostró compromiso de segunda motoneurona. El análisis molecular mostró una expansión anormal de tripletes citosina-adenina-guanina en el gen de receptor de andrógeno en todos los casos. Todos los pacientes cursaron con una presentación clínica típica de la enfermedad siendo los primeros casos de enfermedad de Kennedy con diagnóstico molecular realizado en el Perú.
Palabras clave: Atrofia bulboespinal ligada al X; Receptores androgénicos; Enfermedades genéticas ligadas al cromosoma X (fuente: DeCS BIREME).
Abstract
Kennedy’s disease is an X-linked recessive disorder with onset in adulthood, characterized by progressive degeneration of spinal motor neurons due to a dynamic mutation in the androgen receptor gene. We report three families (five cases) characterized by progressive weakness involving both limbs and bulbar muscles, atrophy, tremor, cramps and endocrinologic disturbances; the neurophysiological studies demonstrated second motor neuron impairment. The molecular analysis identified abnormal CAG repeats expansion in the androgen receptor gene (AR) in all cases. Clinical features were consistent with other previous reports. These are the first Peruvian cases of Kennedy´s disease with confirmed molecular diagnosis.
Key words: Bulbo-spinal atrophy, X-linked; Receptors, androgen; Genetic diseases, X-linked (source: MeSH NLM).
INTRODUCCIÓN
La enfermedad de Kennedy es un trastorno degenerativo de neurona motora espinal, de inicio en la adultez (1) descrita por primera vez en 1966, y cuya mutación genética fue identificada en 1991(2). Debido a su carácter recesivo ligado al cromosoma X son los varones los afectados clínicamente (3); la prevalencia reportada de la enfermedad es 3,3/100 000 habitantes en caucásicos europeos(4).
La enfermedad se origina por una mutación dinámica en el gen de receptor de andrógenos (AR, Xq11-q12), que consiste en una repetición anormalmente expandida en el triplete citosina-adenina-guanina (CAG) en exón 1 del gen. Fenotípicamente, los pacientes cursan con debilidad progresiva en los músculos de las extremidades y bulbares, atrofia, fasciculaciones, temblor, calambres y alteraciones endocrinológicas (2,5).
Se ha descrito afectación sensitiva como hipoestesias y/o parestesias hasta en un tercio de los casos (1). El diagnóstico definitivo de la enfermedad se realiza demostrando la presencia de más de 40 tripletes CAG en el gen AR (3). Se presentan cinco casos de enfermedad de Kennedy en tres familias, con análisis molecular realizado en Perú.
REPORTE DE CASOS
Previo proceso de consentimiento informado, se realizó la evaluación clínica y posterior análisis molecular con muestras de ADN de cinco individuos con sospecha de enfermedad de Kennedy, utilizando la técnica de reacción en cadena de polimerasa (PCR) con termociclador Applied Biosystem 9700 y cebadores específicos: forward (5-TCCAGAATCTGTTCCAGAGCGTGC-3) y reverse (5-GCTGTGAAGGTTGCTGTTCCTCAT-3), electroforesis en gel de poliacrilamida al 6% y tinción del gen con nitrato de plata.
FAMILIA EK1
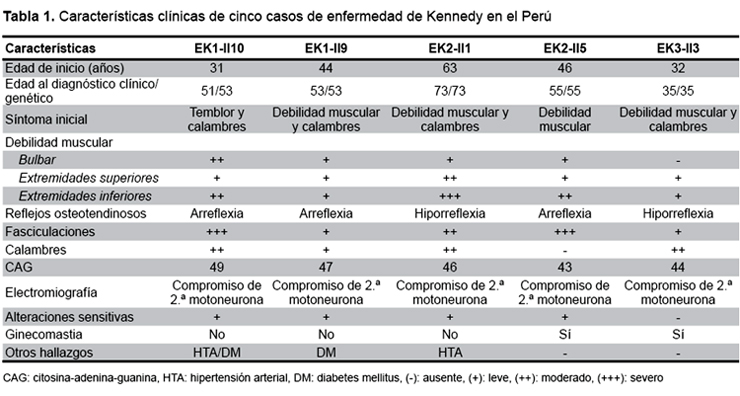
El individuo (II10) es un varón de 51 años, de ascendencia china, con debilidad muscular generalizada de 20 años de evolución lentamente progresiva, agregándose disfagia, disartria, calambres y temblor en el curso de la enfermedad. Examen neurológico: cuadriparesia flácida asimétrica proximal, sin signos piramidales, marcha miopática, hipoestesia distal bilateral, fasciculaciones generalizadas, temblor en manos y compromiso bilateral de los nervios craneales V, IX, X, XI y XII (Tabla 1). Otros hallazgos: microorquidia, vello pubiano escaso; antecedentes: hipertensión arterial y diabetes mellitus de seis años de evolución. Laboratorio: creatina fosfoquinasa 240 UI/mL, el perfil analítico hormonal disponible solo en este caso mostró valores disminuidos de testosterona (2,26 ng/mL) y LH (1,99 mIU/mL). El paciente presentó un episodio de neumonía aspirativa que evolucionó favorablemente. Su hermano mayor (II9), de 53 años presenta diabetes mellitus II y un cuadro clínico similar de nueve años de evolución (Figura 1A).

FAMILIA EK2
El individuo II1 es un varón mestizo de 73 años, con debilidad muscular generalizada progresiva de 10 años de evolución, agregándose calambres, disartria, hipofonía y caídas frecuentes. Examen neurológico: cuadriparesia flácida asimétrica proximal, sin signos piramidales, marcha en estepaje, hipoestesia distal bilateral, fasciculaciones generalizadas y voz nasal. (Tabla_1). Antecedente: hipertensión arterial de larga evolución. Su hermano menor (II5), de 55 años presenta también un cuadro clínico similar, de nueve años de evolución (Figura 1B).
FAMILIA EK3El individuo II3 es un varón mestizo de 35 años, con calambres en miembros inferiores y debilidad muscular generalizada de 3 años de evolución. Examen neurológico: cuadriparesia fláccida asimétrica proximal, fasciculaciones generalizadas, sin signos piramidales ni alteraciones sensitivas (Figura 1C).
DISCUSIÓN
La enfermedad de Kennedy es un trastorno neurodegenerativo asociado a expansión anormal de tripletes CAG en el gen que codifica el receptor de andrógeno (1,6). La reciente implementación del estudio molecular para esta enfermedad en el Perú, ha permitido el diagnóstico genético de los primeros cinco casos en el país.
Esta enfermedad es de presentación tardía; la edad promedio de inicio de síntomas en nuestros casos es 43,2 años (31-63 años), consistente con lo descrito en la literatura, que reporta rangos entre la segunda y sexta década de la vida (2,5).
En cuatro de los casos (EK1-II9, EK2-II1, EK2-II5 y EK3-II3) el síntoma inicial fue la debilidad muscular en miembros inferiores con dificultad para la marcha; el caso restante debutó con temblor postural en mentón y manos, asociándose temblor de voz en el curso de la enfermedad y calambres; esto contrasta con otros reportes donde, usualmente, el temblor es el síntoma más precoz, particularmente en manos y región perioral, aunque asociado a temblor de voz, hasta en el 62% de los casos (1). En menos del 5%, la debilidad muscular es un síntoma inicial (7) aunque es probable que en nuestros casos haya sido considerado como tal por ser el más discapacitante para su desempeño cotidiano.
Los síntomas neurológicos predominantes en los casos fueron cuadriparesia, temblor, calambres, fasciculaciones y compromiso bulbar variable, reconocidos como hallazgos característicos de la enfermedad (2). La cuadriparesia a predominio proximal y asimétrica tuvo un patrón ascendente lentamente progresivo afectando extremidades inferiores, superiores y, finalmente, segmentos bulbares y de músculos faciales como se ha descrito en otros reportes (1). Los individuos EK1-II10, EK2-II1 y EK3-II3 manifestaron calambres que se exacerban con el ejercicio al inicio de la enfermedad, característicos del cuadro clínico (8).
Todos nuestros casos cursaron con fasciculaciones, aunque este síntoma está descrito solo en el 10% de casos (1); estas fasciculaciones fueron generalizadas y comprometieron, inclusive, la lengua y la región perioral incrementado con la contracción voluntaria de los músculos relacionados, como está referido en otros estudios (2,3). El individuo EK3-II3 no presentó síntomas bulbares (disfagia, disartria, disfonía) lo cual se explica por su característica presentación tardía en la enfermedad como reportaron Sperfeld et al. en 100% de su serie (7). La voz nasal, descrita en los casos EK2-II1 y EK2-II5, estaría en relación con la paresia palatina (2).
El probando EK1-II10 presentó parestesias en extremidades inferiores, hipoestesia distal dolorosa, y alteración en la sensibilidad vibratoria, descritas en menos de la mitad de casos en otros estudios (1,9). Si bien está descrito el compromiso de las neuronas sensitivas (2), es difícil atribuir dicho hallazgo únicamente a la enfermedad, ya que el paciente presentaba diabetes mellitus 2.
En todos los casos la electromiografía con aguja mostró actividad espontánea de denervación y reinervación (potenciales de fibrilación, fasciculación y potenciales de acción de unidad motora neurogénicos crónicos de cambios más pronunciados que los potenciales de fibrilación) con compromiso de segunda motoneurona; estos hallazgos se correlacionan con un lento y crónico proceso degenerativo de las células del asta anterior (10). El caso EK1-II10 presentó, además, polineuropatía axonal sensitivo-motora considerado como proceso degenerativo primario en los nervios motores y sensitivos; el componente sensitivo es considerado infrecuente (3).
Los hallazgos no neurológicos de nuestros pacientes incluyeron la presencia de ginecomastia, hipertensión arterial y diabetes mellitus. Los casos EK2-II5 y EK3-II3 presentaron ginecomastia, lo que contrasta con la frecuencia de 70% descrita por Dejager et al. (11), siendo considerada la manifestación no neurológica más frecuente (2). Los dos individuos EK1-II10 y EK2-II1 presentaron hipertensión arterial, y el individuo EK1-II10 cursó, además, con diabetes mellitus, lo que coincide con lo señalado en pequeñas series de casos sin precisarse los mecanismos fisiopatológicos (11,12). Otras alteraciones referidas en las revisiones son: oligospermia, azoospermia e infertilidad (8,12); ninguno de nuestros pacientes cuenta con dichos estudios dado el limitado acceso en nuestro país; sin embargo, los heredogramas muestran descendencia en los individuos afectados.
El perfil analítico hormonal disponible en el caso EK1-II10 mostró dosajes disminuidos de testosterona y LH; sin embargo, pese a que los valores de testosterona pueden ser disminuidos (13) o incrementados (11) no es posible comparar directamente nuestro resultado por ser un dosaje de testosterona total y no fracción libre como en otras series. Los valores de LH referidos en la literatura suelen ser normales (14).
No existe tratamiento establecido salvo el manejo sintomático (2,3). Los calambres suelen responder al magnesio, el baclofeno, la gabapentina, el valproato o la carbamazepina; el temblor responde a fármacos betabloqueadores como el propranolol, y la diabetes e hipertensión a los tratamientos habituales. La sustitución hormonal con testosterona, o análogos de la misma, podrían incluso deteriorar los síntomas. Se han realizado ensayos basados en la deprivación androgénica en ratones con la mutación con fármacos como la leuprorelina, un agonista LHRH, suprimiendo las manifestaciones de deterioro neuromuscular por inhibición de la acumulación tóxica de la proteína mutante AR. Diferentes estudios experimentales en animales comprenden los inhibidores de las desacetilasa de histonas, inhibidores de la Hsp 90 (heat shock protein 90), entre otros. Estos productos no están disponibles para uso en la práctica clínica en el país. El paciente EK1-II10 presentó neumonía aspirativa, de buena evolución, la cual, junto a la falla respiratoria, son las complicaciones y causas de muerte más frecuentemente descritas (2).
La mayoría de casos estarían asociados a un efecto fundador asiático y/o europeo; las neomutaciones son muy raras (15). Una de las familias (EK1) es de origen étnico chino han. Debido al patrón de herencia recesiva ligada al X son afectados los hombres, mientras que las mujeres presentan elevaciones asintomáticas de creatina fosfoquinasa y discretas molestias como temblor o fasciculaciones(3).
En conclusión, presentamos los primeros cinco casos de enfermedad de Kennedy con diagnóstico molecular por PCR cualitativo en nuestro país, con un cuadro clínico típico consistente con otras series. Debe considerarse el diagnóstico de esta entidad en todo varón adulto, con debilidad muscular progresiva y alteraciones endocrinas asociados, con o sin antecedente familiar. Este diagnóstico molecular se encuentra ahora disponible en contexto de investigación, al igual que el protocolo de asesoramiento genético para las personas afectadas y sus familias.
Agradecimientos: al NIH Research Training Grant # R25 TW009345, por apoyo al entrenamiento de investigadores. A los Dres. Cyrus Zabetian e Ignacio F. Mata del Laboratorio de Neurogenética del VA Puget Sound Health Care System, Seattle por proveer cebadores para el análisis molecular
Contribuciones de autoría: VGC, MCO, OA, VM, SL, MF y PM participaron en la concepción y diseño del artículo, el análisis e interpretación de datos, redacción del artículo, revisión crítica del artículo y aprobación de su versión final. VGC, MCO, MF y PM participaron en la recolección y obtención de los resultados. VM, OA y SL contribuyeron en el procesamiento de las muestras.
Fuentes de financiamiento: Instituto Nacional de Ciencias Neurológicas, (presupuesto de Investigación).
Conflictos de interés: los autores declaran no tener conflictos de interés.
Referencias Bibliográficas
1. Lee JH, Shin JH, Park KP, Kim IJ, Kim CM, Lim JG, et al. Phenotypic variability in Kennedy’s disease: implication of the early diagnostic features . Acta Neurol Scand. 2005;112(1):57-63. [ Links ]
2. Finsterer J. Perspectives of Kennedy’s disease . J Neurol Sci. 2010;298(1):1-10. [ Links ]
3. Finsterer J. Bulbar and spinal muscular atrophy (Kennedy’s disease): a review . Eur J Neurol. 2009;16(5):556-61. [ Links ]
4. Guidetti D, Sabadini R, Ferlini A, Torrente I. Epidemiological survey of X-linked bulbar and spinal muscular atrophy, or Kennedy disease, in the province of Reggio Emilia, Italy . Eur J Epidemiol. 2001;17(6):587-91. [ Links ]
5. Greenland KJ, Beilin J, Castro J, Varghese PN, Zajac JD. Polymorphic CAG repeat length in the androgen receptor gene and association with neurodegeneration in a heterozygous female carrier of Kennedy’s disease . J Neurol. 2004;251(1):35-41. [ Links ]
6. Greenland KJ, Zajac JD. Kennedy’s disease: pathogenesis and clinical approaches . Intern Med J 2004;34(5):279-86. [ Links ]
7. Sperfeld AD, Karitzky J, Brummer D, Schreiber H, Haussler J, Ludolph AC, et al X-linked bulbospinal neuronopathy: Kennedy disease . Arch Neurol. 2002;59(12):1921-6. [ Links ]
8. Nance MA. Clinical aspects of CAG repeat diseases . Brain Pathol. 1997;7(3):881-900. [ Links ]
9. Udd B, Juvonen V, Hakamies L, Nieminen A, Wallgren-Pettersson C, Cederquist K, et al. High prevalence of Kennedy’s disease in Western Finland-is the syndrome underdiagnosed? Acta Neurol Scand. 1998;98(2):128-33. [ Links ]
10. Ferrante MA, Wilbourn AJ. The characteristic electrodiagnostic features of Kennedy’s disease . Muscle Nerve. 1997;20(3):323-9. [ Links ]
11. Dejager S, Bry-Gauillard H, Bruckert E, Eymard B, Salachas F, LeGuern E, et al. A comprehensive endocrine description of Kennedy’s disease revealing androgen insensitivity linked to CAG repeat length . J Clin Endocrinol Metab. 2002;87(8):3893-901. [ Links ]
12. Atsuta N, Watanabe H, Ito M, Banno H, Suzuki K, Katsuno M, et al. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients . Brain. 2006;129(6):1446-55. [ Links ]
13. Hausmanowa-Petrusewicz I, Borkowska J, Janczewski Z. X-linked adult form of spinal muscular atrophy . J Neurol. 1983;229(3):175-88. [ Links ]
14. Mariotti C, Castellotti B, Pareyson D, Testa D, Eoli M, Antozzi C, et al. Phenotypic manifestations associated with CAG-repeat expansion in the androgen receptor gene in male patients and heterozygous females: a clinical and molecular study of 30 families. Neuromuscul Disord. 2000;10(6):391-7. [ Links ]
15. Tanaka F, Doyu M, Ito Y, Matsumoto M, Mitsuma T, Abe K, et al. Founder effect in spinal and bulbar muscular atrophy (SBMA) . Hum Mol Genet. 1996;5(9):1253-7. [ Links ]
Correspondencia: Víctor Gómez-Calero.
Dirección: Jr. Ancash 1271, Barrios Altos, Lima 1, Perú.
Teléfono: 411-7779.
Correo electrónico: peru.neurogenetica@gmail.com
Recibido: 28-01-13
Aprobado: 03-04-13
