










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
versión impresa ISSN 1726-4634
Rev. perú. med. exp. salud publica vol.31 no.1 Lima ene./dic. 2014
Artículo Original
Validación de un método de cromatografía líquida para la determinación de rifampicina en plasma humano
Validation of a liquid chromatography method for rifampicin determination in human plasma
Luis Moreno-Exebio1,a, Miguel Grande-Ortiz1,a
1 Centro Nacional de Control de Calidad, Instituto Nacional de Salud. Lima, Perú
a Químico Farmacéutico
RESUMEN
Objetivos: Validar un método de cromatografía líquida de alta resolución (HPLC) para la determinación de rifampicina (RFP) en plasma humano. Materiales y métodos. Se desarrolló un método HPLC para la determinación de RFP en plasma. La separación fue realizada por cromatografía de fase reversa con una columna C18 y una fase móvil compuesta por una mezcla de acetonitrilo y solución amortiguadora de fosfato de potasio monobásico 0,05 M (38:62 v/v) a 335 nm. En el cual se empleó como estándar interno rifampicina quinona (RFP-QN). Resultados. Los tiempos de retención de RFP y RFP-QN fueron 7,81 y 12,26 minutos, respectivamente. El ensayo fue lineal de 0,5 a 250 ug/mL Los parámetros evaluados de precisión, exactitud, selectividad, linealidad, recuperación cumplieron con lo establecido en las guías internacionales de validación de métodos bioanalíticos. Conclusiones. El método HPLC desarrollado es simple, específico, sensible, selectivo y lineal para un amplio rango de concentraciones de RFP en plasma.
Palabras clave: Cromatografía líquida de alta presión; Estudios de validación; Rifampicina (fuente: DeCS BIREME)
ABSTRACT
Objectives: To validate the high-performance liquid chromatography method (HPLC) for rifampicin (RFP) determination in human plasma. Materials and methods. A HPLC method for RFP determination in plasma was developed. The separation was performed by reversed-phase chromatography with C18 column and a mobile phase composed of a mixture of acetonitrile and monobasic potassium phosphate buffer solution 0.05 M (38:62 v/v) at 335 nm in which standard rifampicin quinone (RFP-QN) was used. Results. The retention times of RFP and RFP-QN were 7.81 and 12.26 minutes, respectively. The trial was linear from 0.5 to 250 ug/mL. The evaluated parameters of precision, accuracy, selectivity, linearity, and recovery complied with the established international standards for validation of bioanalytical methods. Conclusions. The developed HPLC method is simple, specific, sensitive, selective and linear for a wide range of RFP concentrations in plasma.
Key words: Chromatography, high pressure liquid; Validation studies; Rifampin (source: MeSH NLM).
INTRODUCCIÓN
El monitoreo fármaco terapéutico (MFT) es el proceso de obtener las concentraciones plasmáticas de un fármaco y modificar la dosis basados en los resultados y en la evaluación clínica para optimizar los beneficios terapéuticos, mientras se minimizan sus riesgos por efectos adversos o toxicidad. El monitoreo terapéutico ha sido aceptado para un grupo grande de fármacos, incluyendo los usados en el tratamiento de la tuberculosis (1). Al igual que en otras enfermedades, el uso de MFT en tuberculosis permite a los clínicos tomar decisiones informadas respecto al ajuste de la dosis en terapia. En otras palabras, el MFT puede ayudar a identificar que pacientes requieren mayores dosis (2,3).
La rifampicina (RFP), así como la isoniacida (INH), causan la mayor y más temprana reducción en el número de Mycobacterium tuberculosis; es por ello que ambas drogas son consideradas como los dos principales fármacos para combatir la tuberculosis y forma parte fundamental de los regímenes de primera línea antituberculosos en el Perú y a nivel mundial (4,5).
Se ha descrito que bajas concentraciones de RFP en el plasma han sido asociadas con falla al tratamiento contra la tuberculosis (6). Es así que, la detección temprana de personas que no alcanzan concentraciones plasmáticas adecuadas de RFP o INH, permitiría realizar un oportuno ajuste de dosis o un cambio de esquema de tratamiento. Tales ajustes no se requieren en aquellos pacientes que responden al régimen estándar de cuatro fármacos. No obstante, algunos pacientes que responden lentamente al tratamiento, que tienen una mala absorción, están en riesgo de interacciones medicamentosas o tienen otras enfermedades concomitantes que complican la situación clínica, podrían beneficiarse del MFT con intervenciones oportunas que pueden evitar el desarrollo de resistencia (7).
Existen, sin embargo, pocos métodos sencillos y rápidos para determinar la concentración plasmática de RFP en pacientes, lo son mucho menos los que emplean cromatografía líquida de alta resolución (HPLC). La mayoría de métodos existentes demandan procedimientos tediosos o requieren equipamiento especial (8). Es por ello que el objetivo de este estudio fue estandarizar un método para determinar la concentración de RFP en el plasma, modificando ciertas condiciones experimentales de los métodos ya existentes para usarlo en el monitoreo terapéutico de pacientes del programa de tuberculosis del Ministerio de Salud del Perú (MINSA).
MATERIALES Y MÉTODOS
SISTEMA CROMATOGRÁFICO
Se empleó un sistema HPLC (LaChrom Elite® HPLC System), el cual estuvo conformado por: una bomba (L-2130), un detector de arreglo de diodos (L-2455), un automuestreador con sistema de enfriamiento (L-2200) y un horno para columna (L-2350). El software para la recolección y procesamiento de la información fue EZ Chrom Elite® versión 3.2.1 (Cromatography Data System).
La columna utilizada fue C18 (PurosphereStar®) 150 x 4,6 mm ID, el tamaño de partícula fue de 5 um (Merck KGaA, Darmstadt, Germany), con una precolumna 4-mm L X 3,0 mm ID (Lichrospher 100 RP-18e, 5 um, Merck KGaA, Darmstadt, Germany). La fase móvil estuvo compuesta por una mezcla de acetonitrilo y solución amortiguadora 0,05 M de fosfato de potasio monobásico (0,05 mol/L de fosfato de potasio monobásico KH2PO4 ajustado a pH 3,7 con ácido fosfórico) en una proporción 38:62 v/v, con una velocidad de flujo de 1 mL/min a temperatura ambiente. El volumen de inyección y la longitud de onda de cuantificación fueron 20 uL y 335 nm respectivamente. Las muestras se mantuvieron en el automuestreador a 5 ºC durante todo el análisis.
REACTIVOS
Se utilizaron estándares primarios de RFP y rifampicina quinona (RFP-QN) de la United States Pharmacopeia, acetonitrilo (J.T. Baker, USA), metanol grado HPLC (J.T. Baker, México). Además, se emplearon fosfato de potasio monobásico, y ácido fosfórico (Merck KGaA, Darmstadt, Germany) de grado analítico. Asimismo, se usó agua grado HPLC (18.2 MΩ), obtenida a través de un equipo purificador de agua Sartorius Stedim Biotech modelo Arium ® 611 UV (Gottingen, Germany).
PREPARACIÓN DE LA SOLUCIÓN ESTÁNDAR
Se preparó una solución madre de 1000 ug/mL de RFP en metanol. Diferentes volúmenes de la solución madre de RFP se diluyeron en metanol y plasma para producir los estándares de calibración de RFP. Una solución del estándar interno de RFP-QN, se diluyó con metanol a partir de una solución madre para obtener una concentración final de 100 ug/mL. Todas las soluciones fueron almacenadas en tubos de polipropileno a -70 °C. Se prepararon estándares de calibración de RFP a concentraciones de 0,5; 1, 5; 10; 20; 50; 100; 200 y 250 ug/mL añadiendo diferentes volúmenes de solución madre de RFP y 100 uL de solución del estándar interno en 5 mL de plasma.
Se utilizó muestras de control de calidad (McC) (9,10) en plasma a concentraciones: baja (1 ug/mL), intermedia (10 ug/mL) y alta (20 ug/mL). Los estándares de calibración y de control de calidad fueron extraídos cada día de análisis con el procedimiento usado para la preparación de muestras.
PREPARACIÓN DE MUESTRAS
En crioviales de 2,0 mL que contienen 100 uL de estándar interno (100 ug/mL) se adicionó 100 uL de cada uno de los estándares de calibración y 100 uL de acetonitrilo, después de mezclar en un vórtex a 2200 rpm por 3 min, se centrifugó a 8000 rpm por 10 min; se inyectó 20 uL del sobrenadante, en el sistema de HPLC. De la misma manera, se preparó un blanco (plasma sin estándar interno) y un blanco cero (plasma más estándar interno).
VALIDACIÓN DEL MÉTODO
Se tomaron en cuenta los parámetros recomendados por la guía de la Food and Drug Administration (FDA) para la validación de métodos bioanalíticos (9) y la guía de la European Medicines Agency (EMA) (10). En las cuales se señala que los parámetros principales de validación son la selectividad, exactitud, precisión, recuperación, curva de calibración y estabilidad. Para la evaluación de estos parámetros se tuvieron las siguientes consideraciones:
Selectividad. Se preparó una solución estándar de isoniazida, pirazinamida y etambutol de 40 ug/mL en metanol; inyectándose 20 uL de esta solución en el equipo HPLC. Estos medicamentos se seleccionaron porque se utilizan conjuntamente con la RFP para tratar a los pacientes en el esquema de primera línea del programa de tuberculosis del MINSA.
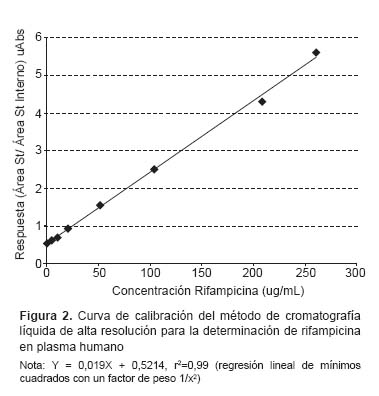
Linealidad. Se preparó una curva de calibración analizando los estándares de RFP de 9 concentraciones (0,5–250 ug/mL), usando para su evaluación una regresión lineal de mínimos cuadrados.
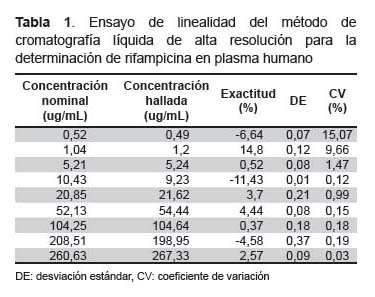
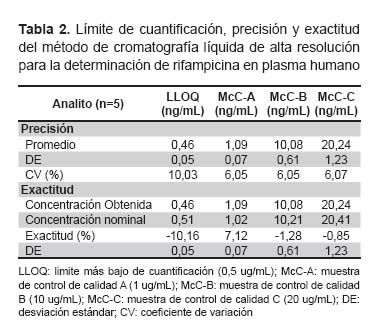
Exactitud y precisión. Se realizó cinco inyecciones consecutivas del límite más bajo de cuantificación (0,5 ug/mL) y de las McC. La exactitud fue evaluada a través de la desviación del promedio obtenido de las concentraciones 0,5 (es decir, del límite más bajo de cuantificación), 1, 10 y 20 ug/mL (es decir de las McC) en relación a la concentración real del analito. La precisión intradía del método es determinada a través del coeficiente de variación de cada concentración, el cual no debe exceder de 20% para las muestras correspondientes al límite más bajo de cuantificación (LLOQ) y 15% para las McC. El mismo procedimiento se repitió durante cinco días consecutivos para determinar la precisión interdía.
Recuperación. Se preparó cinco inyecciones de las McC en plasma y en fase móvil y se comparó sus concentraciones. El límite más bajo de cuantificación (LLOQ) fue definido como la concentración más baja sobre la curva de calibración con una precisión de <20% y una exactitud ± 20%.
RESULTADOS
En relación a la selectividad se observó que, aun cuando fueron analizadas bajo las mismas condiciones cromatográficas que las soluciones de RFP y RFP-QN, ninguna de las soluciones estándar de isoniazida, pirazinamida y etambutol (Figura 1) presentan señal que pueda interferir con el tiempo de retención de la RFP (7,8 min) y RFP-QN (12,3 min), conforme se aprecia en la Figura 1
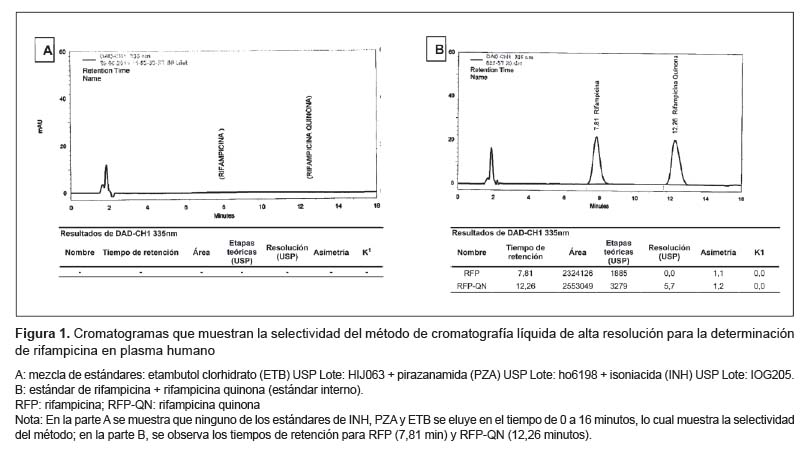
El ensayo de linealidad fue medido por un análisis de regresión lineal de mínimos cuadrados con un factor de peso 1/x2 (Tabla 1). La curva de calibración (Figura 2) de RFP fue y = 0,019X + 0,5214 (r2=0,99), correspondiendo al rango de 0,5-250 ug/mL. El LLOQ obtenido con la concentración más baja (0,5 ug/mL) sobre la curva de calibración mostró una precisión de 10% (C.V.) y una exactitud ± 10%.
El sistema de precisión fue evaluado por área del pico de RFP versus el estándar interno después de cinco inyecciones sucesivas de los estándares de control de calidad (McC). Los valores obtenidos de LLOQ y McC se presentan en la Tabla 2.
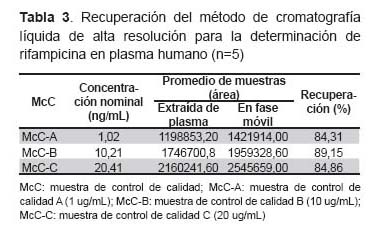
La recuperación promedio alcanzada con este método fue de 86,1%. Los valores obtenidos para cada uno de los puntos de control de calidad se muestran en la Tabla 3.
DISCUSIÓN
El método de cromatografía líquida para la determinación de RFP en plasma humano utilizado en la presente investigación demostró ser bastante selectivo. La corrida cromatográfica (16 min), permitió una selectividad adecuada de la RFP respecto a los otros fármacos de primera línea empleados en el tratamiento antituberculoso. Se puede afirmar que este método es comparable con la separación obtenida con métodos más sensibles, como la cromatografía líquida acoplada a un espectrómetro de masa (HPLC-MS) (11).
Los métodos existentes para la determinación de RFP en muestras biológicas han sido validados en un rango de concentraciones que varían generalmente entre 0,4-20 ug/mL (12), otros entre 0,25 y 15 ug/mL (8). En algunos trabajos recientes se encontró valores por encima de 20 ug/mL (13); siendo RFP el fármaco con el más alto potencial de variabilidad entre los fármacos antituberculosos (1,14); por esta razón en el presente trabajo se amplió el rango de la curva de calibración de 0,5 hasta 250 ug/mL.
La exactitud hallada estaba dentro de los valores recomendados por la guía de validación de la FDA y EMA, que indica: el valor promedio de McC deben estar dentro del 15% del valor verdadero y 20% para LLOQ. Asimismo, la precisión obtenida cumple lo recomendado, el coeficiente de variación no excede el 15% para las McC y el 20% para LLOQ. Esta exactitud y precisión fue comparable a otros métodos de HPLC (15). El valor promedio de recuperación del método encontrado en el presente estudio fue 86%, el cual es muy similar al encontrado por Zhou et al. realizado en plasma de pacientes chinos (16) que alcanzó valores superiores al 83%.
La estabilidad de corto plazo de rifampicina en el plasma fue evaluada comparando las áreas de los McC durante las pruebas de validación, sin hallar variación respecto al área de RFP o RFP-QN a lo largo de corridas cromatográficas de más de 48 horas porque la bandeja del automuestreador se programó a 5 ºC.
Una ventaja del método propuesto es que este usa un estándar interno (RFP-QN) de fácil adquisición y bajo costo, en comparación con otros métodos para la cuantificación de RFP que utilizan estándares internos de difícil adquisición por ser sustancias controladas (5), o que usan metabolitos activos de costo elevado (17,18). Además, la RFP-QN es empleada en la determinación de contenido de RFP en capsulas de la farmacopea de Estados Unidos - USP 35 (19).
Una limitación debe ser reconocida, la duración de la corrida cromatográfica es relativamente larga en comparación con otros métodos de HPLC-MS. Sin embargo, el método propuesto es simple, preciso y exacto y constituye una buena alternativa para pruebas de monitoreo terapéutico de RFP en el tratamiento de pacientes con tuberculosis.
En conclusión, el método de cromatografía líquida para la determinación de rifampicina en plasma humano, validado en el presente trabajo constituye una herramienta valiosa. Su uso conjunto con los datos clínicos y bacteriológicos del paciente permitiría tratar los casos más complicados de tuberculosis de una manera adecuada. La validación de este método de cromatografía líquida de alta resolución posibilitaría su implementación como parte del MFT de pacientes del programa nacional de tuberculosis en Perú que se encuentren en tratamiento con drogas de primera línea. Gracias a este metodo se podría detectar de forma temprana las concentraciones plasmáticas subterapéuticas de RFP en pacientes que se encuentren en mayor riesgo de fracaso al tratamiento (20). Se requieren de estudios costo-beneficio para evaluar la factibilidad de su implementación en hospitales del MINSA.
Agradecimientos: al Dr. Carlos Coimbra, del Instituto Fio Cruz de Brasil, por la el apoyo y las sugerencias durante la preparación del artículo.
Contribuciones de autoría: LME participó en la concepción y diseño del estudio, en la recolección, análisis e interpretación de datos, en la redacción del artículo. MGO ha participado en el análisis e interpretación de datos y revisión crítica del artículo. Ambos autores aprobaron la versión final a publicar.
Fuentes de financiamiento: el presente trabajo fue realizado con fondos provenientes del Instituto Nacional de Salud.
Conflictos de interés: los autores declaran no tener conflictos de interés.
Referencias Bibliográficas
1. Peloquin CA. Therapeutic drug monitoring in the treatment of tuberculosis. Drugs 2002;62(15):2169-83. [ Links ]
2. Kang JS, Lee MH. Overview of therapeutic drug monitoring. Korean J Intern Med. 2009;24(1):1-10. doi: 10.3904/kjim.2009.24.1.1. [ Links ].
3. Requena-Méndez A, Davies G, Ardrey A, Jave O, López-Romero SL, Ward SA, et al. Pharmacokinetics of rifampicin in Peruvian tuberculosis patients with and without comorbid diabetes or HIV. Antimicrob. Agents Chemother. 2012;56(5):2357-63. doi: 10.1128/AAC.06059-11. [ Links ]
4. Perú, Ministerio de Salud. Norma Técnica de Salud para el Control de la Tuberculosis. NTS. Nº 041-MINSA/DGSP – V.01. Lima: MINSA; 2006. [ Links ]
5. Zhang Y. Advances in the treatment of tuberculosis. Clin Pharmacol Ther. 2007;82(5):595-600. [ Links ]
6. Mehta JB, Shantaveerapa H, Byrd RP Jr, Morton SE, Fountain F, Roy TM. Utility of rifampin blood levels in the treatment and follow-up of active pulmonary tuberculosis in patients who were slow to respond to routine directly observed therapy. Chest. 2001;120(5):1520-4. [ Links ]
7. Peloquin C. Use of therapeutic drug monitoring in tuberculosis patients. Chest. 2004;126(6):1722-4. [ Links ]
8. Hemanth Kumar AK, Chandra I, Geetha R, Chelvi KS, Lalitha V, Prema G. A validated high-performance liquid chromatography method for the determination of rifampicin and desacetyl rifampicin in plasma and urine. Indian J Pharmacol. 2004;36(4):231-3. [ Links ]
9. Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for Industry. Bioanalytical Method Validation. Rockville: FDA; 2001. [ Links ]
10. European Medicine Agency (EMA). Guideline on bioanalytical method validation. EMEA/CHMP/EWP/192217/2009. London: EMA; 2011. [ Links ]
11. Song SH, Jun SH, Park KU, Yoon Y, Lee JH, Kim JQ, et al. Simultaneous determination of first-line anti-tuberculosis drugs and their major metabolic ratios by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2007;21(7):1331-8. [ Links ]
12. Smith PJ, van Dyk J, Fredericks A. Determination of rifampicin, isoniazid and pyrazinamide by high performance liquid chromatography after their simultaneous extraction from plasma. Int J Tuberc Lung Dis. 1999;3(11 Suppl 3):S325-8 [ Links ]
13. Heysell SK, Moore JL, Keller SJ, Houpt ER.. Therapeutic drug monitoring for slow response to tuberculosis treatment in a state control program, Virginia, USA. Emerg Infect Dis. 2010;16(10):1546-53. doi: 10.3201/eid1610.100374. [ Links ]
14. Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352(21):2211-21. [ Links ]
15. Allanson AL, Cotton MM, Tettey JN, Boyter AC. Determination of rifampicin in human plasma and blood spots by high performance liquid chromatography with UV detection: a potential method for therapeutic drug monitoring. J Pharm Biomed Anal. 2007;44(4):963-9. [ Links ]
16. Zhou Z, Chen L, Liu P, Shen M, Zou F. Simultaneous determination of Isoniazid, Pyrazinamide, Rifampicin and Acetylisoniazid in Human Plasma by high performance liquid chromatography. Anal Sci. 2010;26(11):1133-8. [ Links ]
17. Um SW, Lee SW, Kwon SY, Yoon HI, Park KU, Song J, et al. Low serum concentration of anti-tuberculosis drug and determinants of serum levels. Int J Tuberc Lung Dis. 2007;11(9):972-8. [ Links ]
18. Prakash J, Velpandian T, Pande JN, Gupta SK. Serum rifampicin levels in patients with tuberculosis: effect of P-Glycoprotein and CYP3A4 blockers on its absorption. Clin Drug Investig. 2003;23(7):463-72. [ Links ]
19. Farmacopea de los Estados Unidos USP 35. Maryland: United Book Press Inc.; 2012. [ Links ]
20. Ray J, Gardiner I, Marriott D. Managing antituberculosis drug therapy by therapeutic drug monitoring of rifampicin and isoniazid. Intern Med J. 2003;33(5-6):229-34. [ Links ]
Correspondencia: Luis Moreno Exebio
Dirección: Av. Defensores del Morro 2268 – Chorrillos
Teléfono: 6176200- Anexo 1524 – Celular: 996728185
Correo electrónico: lemoreno70@hotmail.com / lmoreno@ins.gob.pe
Recibido: 16-09-13
Aprobado: 08-01-14

