










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
versión impresa ISSN 1726-4634
Rev. perú. med. exp. salud publica vol.31 no.2 Lima abr. 2014
Reporte de Caso
Enfermedad de creutzfeldt-jakob en el Perú: reporte de once casos
Creutzfeldt-jakob disease in Peru: report of eleven cases
Luis Torres-Ramírez1,a, Jorge Ramírez-Quiñones1,b, Carlos Cosentino-Esquerre1,a, Miriam Vélez-Rojas 1,a, Martha Flores-Mendoza1,a, Diana Rivas-Franchini2,c, Rafael Suarez-Reyes1,a, Yesenia Núñez-Coronado1,a
1 Departamento de Enfermedades Neurodegenerativas, Instituto Nacional de Ciencias Neurológicas. Lima, Perú.
2 Departamento de Neuropatología, Instituto Nacional de Ciencias Neurológicas. Lima, Perú.
a Médico neurólogo; b médico residente de neurología; c médico patólogo.
RESUMEN
La enfermedad de Creutzfeldt-Jakob (ECJ) es una enfermedad neurológica fatal producida por la isoforma patológica de la proteína priónica humana. Se reporta las características clínicas de seis casos de la forma esporádica de ECJ con diagnóstico definitivo por histopatología, y cinco casos con diagnóstico probable, en pacientes atendidos en el Instituto Nacional de Ciencias Neurológicas del Perú. La edad de inicio en los casos definitivos fue de 55,8 años y, en los probables, de 59,6 años, con predominio del sexo masculino. El tiempo de enfermedad fue de 8,8 meses. Se encontró un EEG típico en 50% de los casos definitivos y 80% de los probables. La proteína 14-3-3 en líquido cefalorraquídeo fue positiva en un caso probable y los hallazgos típicos en resonancia magnética se observaron en dos casos probables. Todos los casos cursaron con una evolución clínica típica de la enfermedad, y se considera el primer reporte de ECJ en el Perú.
Palabras clave: Síndrome de Creutzfeldt-Jakob; Enfermedades por prión; Proteínas PrPSc; Priones; Perú (fuente: DeCS BIREME).
ABSTRACT
Creutzfeldt-Jakob disease (CJD) is a fatal neurological disease caused by pathological isoform of the human prion protein. Clinical features of six cases of the sporadic form of CJD with definitive diagnosis by histopathology, and five cases with probable diagnosis were reported in patients treated at the Peruvian National Institute of Neurological Sciences. The average age of onset in definite cases was 55.8 years and in probable cases was 59.6, mostly males. The average disease duration was 8.8 months. A typical EEG was found in 50% of definite cases and in 80% of probable. The 14-3-3 protein in cerebrospinal fluid was positive in a probable case, and typical MRI findings were observed in two probable cases. All cases studied had a typical clinical course of the disease, and it is considered as the first report of CJD in Peru.
Key words: Creutzfeldt-Jakob syndrome; Prion diseases; PrPSc proteins; Prions; Peru (source: MeSH NLM).
INTRODUCCIÓN
La enfermedad de Creutzfeldt-Jakob (ECJ) es la enfermedad por priones más común en humanos (1). Fue descrita por Hans Creutzfeldt en 1920 y Alfons Jakob, un año después, en cinco pacientes con características clínicas heterogéneas bajo el nombre de seudoesclerosis espástica. Walther Spielmeyer, en 1922, acuñó el término de ECJ (2). Es producida por la presencia en el parénquima cerebral de la isoforma patológica de la proteína priónica (PrPsc) la cual resulta del plegamiento anormal de la isoforma normal (PrPc) con mayor contenido de estructura de lámina beta y, por consiguiente, mayor resistencia a la digestión proteolítica y mayor agregación intracelular (3,4).
La ECJ se manifiesta característicamente por una demencia rápidamente progresiva y otros signos neurológicos (1). Existen cuatro formas etiológicas de la enfermedad en humanos: la forma esporádica (esECJ), familiar (fECJ), iatrogénica (iECJ) y la variante (vECJ), cada una con características particulares. La esECJ es la más común, representa alrededor del 85% de los casos de ECJ, con una incidencia anual de 1 caso/millón habitantes (5,6) y es producida por la generación endógena de PrPsc debido a plegamientos anormales aleatorios en la estructura de la PrPc, con mayor frecuencia en individuos con predisposición genética (4,7,8). El diagnóstico definitivo de esECJ se realiza mediante el estudio histopatológico; aunque mediante pruebas diagnósticas como el electroencefalograma, el análisis del líquido cefalorraquídeo y la resonancia magnética, se puede realizar un diagnóstico probable.
En el presente artículo se reportan las características clínicas de seis casos con diagnóstico definitivo por confirmación histopatológica y se describen los hallazgos en cinco casos probables de esECJ atendidos entre los años 1998 y 2013 en el Departamento de Enfermedades Neurodegenerativas del Instituto Nacional de Ciencias Neurológicas del Perú.
REPORTE DE CASOS
CASO 1
Varón de 64 años. Inicia con visión borrosa, mareos, inestabilidad para la marcha, alteración de la memoria e insomnio. Al examen se le encontró parcialmente orientado, con hemiparesia derecha y espasticidad, hiperreflexia y signo de Babinski presente, rigidez en hemicuerpo izquierdo, dismetría bilateral; apraxia y compromiso de memoria. En las semanas siguientes presentó mioclonías periorales y en miembros superiores, deterioro rápidamente progresivo de funciones superiores, signo del miembro extraño en miembro superior izquierdo (MSI), con postración ulterior. Falleció a los 9 meses de enfermedad. El estudio histopatológico mostró abundantes vacuolas en el neuropilo de la corteza cerebral, el cerebelo y los núcleos basales, asociado a gliosis extensa, compatibles con ECJ. No se realizó inmunomarcación de PrPsc.
CASO 2
Varón de 35 años. Tiempo de enfermedad de un año; caracterizado por ánimo depresivo, por lo que recibe fluoxetina. Meses después se añadió alteración de memoria (corto y largo plazo) y fallas en la escritura (escritura incorrecta de algunas palabras). Progresó con inestabilidad para la marcha, olvidó la utilidad de las cosas y presentó pérdida del cuidado personal. En la evaluación se le encontró bradipsíquico, desorientado, con conducta pueril, rigidez en hemicuerpo izquierdo, posturas distónicas, mioclonías generalizadas, presencia de reflejo de prehensión y palmomentoniano; fluencia verbal disminuida, repetición y nominación alteradas; apraxia ideatoria. En las semanas siguientes: cuadriparesia espástica hiperrefléxica con signo de Babinski bilateral llegando a estado de mutismo aquinético. Falleció a los 15 meses de enfermedad. Estudio histopatológico: marcada vacuolización neuronal asociada a gliosis reactiva en corteza y núcleos basales con distribución irregular.
CASO 3
Mujer de 64 años. Dos meses antes del ingreso presentó temblor de miembro superior derecho, inestabilidad para la marcha, alucinaciones visuales, dificultad para nominar objetos y labilidad emocional. Al examen, se encontró desorientada, con hemiparesia derecha espástica hiperrefléxica, mioclonías generalizadas a predominio de miembros superiores; en la evaluación de funciones superiores: afasia global. Cursó con evolución tórpida con crisis tónico-clónicas generalizadas y deterioro del nivel de conciencia. Falleció a los 6 meses de enfermedad. El estudio histopatológico evidenció gliosis reactiva con presencia de astrocitos gemistocíticos y moderada vacuolización neuronal en diferentes zonas del cerebro a predominio frontal.
CASO 4
Varón de 59 años. Tiempo de enfermedad de dos semanas; caracterizado por visión borrosa, inestabilidad para la marcha, habla incoherente e incontinencia urinaria. En la evaluación se encontró paciente parcialmente orientado, bradipsíquico, hipoactivo, con espasticidad generalizada; en la evaluación de funciones superiores: apraxia, agnosia, compromiso de memoria a corto plazo. Cursó con deterioro rápidamente progresivo de funciones superiores, presentó mioclonías generalizadas y evolucionó a estado de mutismo aquinético. Falleció a los 4 meses de enfermedad. Estudio histopatológico: en la corteza cerebral, el cerebelo y los núcleos basales, abundantes vacuolas de 5-10 micras a nivel del neuropilo, acompañada de abundante gliosis que reemplaza parénquima normal. Escasas neuronas de aspecto encogido y citoplasma eosinófilo intenso.
CASO 5
Varón de 69 años. Tiempo de enfermedad de cuatro semanas; caracterizado por insomnio, no sabía el nombre de los objetos, olvidó la forma de comer y vestirse, habla incoherente, episodios de autoagresión y desorientación. Al examen, se encontró paciente parcialmente orientado, hipoactivo, con temblor de reposo y postural en miembros superiores. Durante las semanas siguientes el cuadro progresó con hemiparesia derecha espástica hiperrefléxica, limitación en la supraversión de la mirada, afasia global y mioclonías periorales y en miembros superiores. Falleció a los 5 meses de enfermedad. Estudio histopatológico: corteza cerebral, cerebelo y núcleos basales con presencia focal de espongiosis de neuropilo, marcada gliosis, con astrocitos gemistocíticos y marcada ausencia neuronal.
CASO 6
Mujer de 54 años. Inició con temblor de reposo en miembro superior derecho, apatía, pérdida del cuidado personal, lenguaje incoherente, alucinaciones visuales y pérdida progresiva de la fluencia verbal. Al examen, se encontró desorientada, hipoactiva, con posturas distónicas episódicas, cuadriparesia hipertónica normorrefléxica, mioclonías generalizadas; a la evaluación de funciones superiores: afasia global. Semanas después entró en estado de mutismo aquinético. Falleció a los 13 meses de enfermedad. Estudio histopatológico: focos de espongiosis de neuropilo en corteza cerebral, cerebelo y núcleos basales, definida por vacuolas ovales y redondas de 5-10 micras acompañadas de pérdida neuronal severa y algunos astrocitos gemistocíticos.
DISCUSIÓN
La esECJ es de inicio tardío; la edad promedio de inicio en los casos reportados como definitivos fue de 55,8 años (35-64 años) y en los probables fue de 59,6 años (55-66 años), consistente con otros reportes que indican un pico de edad de inicio entre 55-75 años y una edad promedio de 61 años. De forma infrecuente se presenta en menores de 40 años (9), como en el caso 2. El 64% de los casos reportados fueron de sexo masculino aunque la esECJ afecta en igual proporción a ambos sexos (1).
La tercera parte de los pacientes con esECJ inicia la enfermedad con alteraciones cognitivas y/o conductuales; otro tercio inicia con signos de focalización neurológica (pérdida de la visión, ataxia cerebelosa, afasia o déficit motor), y un tercio restante con síntomas inespecíficos como: fatiga, disminución de apetito, alteraciones del sueño o del humor (1,4). Cuatro de los casos definitivos tuvieron signos neurológicos focales como forma de presentación (visión borrosa y temblor) y dos presentaron insomnio y alteración del humor. En los casos probables, dos iniciaron con signos neurológicos focales, dos con alteraciones del sueño y del humor y uno con compromiso cognitivo. Posteriormente, la mayoría de pacientes con esECJ desarrolla una demencia rápidamente progresiva, disfunción cerebelosa, alteraciones visuales como alucinaciones, agnosia visual y ceguera cortical; y mioclonías espontáneas o inducidas las que suelen estar presentes en alrededor del 90% de casos en fases finales de la enfermedad. El 91% de los casos cursó con mioclonías y el 73% con compromiso visual y/o cerebeloso.
Al igual que en todos nuestros casos, más del 70% individuos con esECJ presentan signos de disfunción piramidal y extrapiramidal (hiperreflexia, espasticidad, rigidez, temblor) y síntomas de depresión (10) durante el curso de la enfermedad. Dos casos presentaron crisis convulsivas, manifestación reportada de forma infrecuente (11). Seis casos (tres definitivos y tres probables) presentaron al final de la enfermedad un estado de mutismo aquinético, dato consistente con la literatura que reporta cifras entre el 30 y el 100% (10). El caso 1 cursó con el signo del miembro extraño en el MSI, el cual es un signo infrecuente, que afecta con mayor frecuencia los miembros del lado izquierdo y puede ser el síntoma inicial de la enfermedad (12). Esta heterogeneidad fenotípica en la forma de presentación y en el curso del esECJ ha sido relacionada a los diversos polimorfismos en el codón 129 del gen PRNP y a las características fisicoquímicas de la PrPsc (13).
El tiempo total de enfermedad (hasta la muerte) es más corto en los pacientes con esECJ comparado con la vECJ. Aproximadamente el 90% de casos de esECJ fallece antes del primer año de enfermedad (1). El promedio en nuestros casos definitivos fue de 8,6 meses y en los probables fue 9 meses, cifras ligeramente mayores a las encontradas en la literatura que las ubican en un rango de 5 a 8 meses en los casos de esECJ. Dicho hallazgo se debe a la presencia de fenotipos no clásicos entre nuestros casos, en los cuales según la literatura el promedio de duración de enfermedad es entre 6 a 17 meses según el polimorfismo encontrado (4).
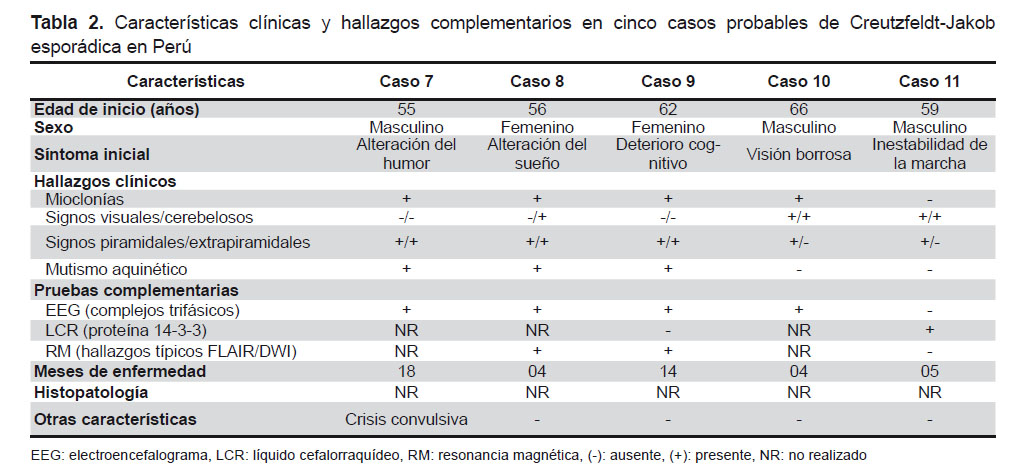
El diagnóstico probable de esECJ según los criterios actuales propuestos por Zerr et al. requiere la presencia de un síndrome demencial rápidamente progresivo y dos signos clínicos (mioclonías, disturbios visuales y/o cerebelosos, signos piramidales/extrapiramidales y mutismo aquinético) asociados a hallazgos típicos en por lo menos una prueba complementaria y la ausencia de un diagnóstico alternativo. Las tres pruebas complementarias son: el electroencefalograma (EEG), el análisis del líquido cefalorraquídeo (LCR) y la resonancia magnética (RM) cerebral (4,11,14). Según el estudio de Zerr et al., la combinación de los resultados de estas pruebas alcanza una sensibilidad y especificidad de 98 y 71% respectivamente (14). Las características clínicas y los hallazgos en las pruebas complementarias de nuestros casos definitivos y probables están descritos en la Tabla 1 y 2 respectivamente.
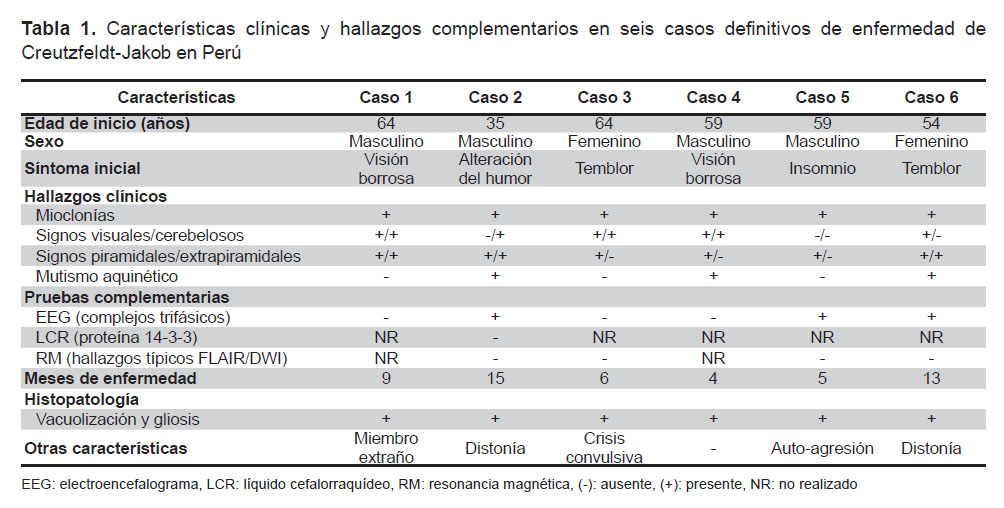
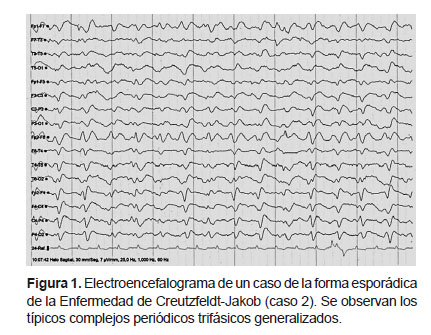
En la forma esECJ, el EEG típicamente muestra complejos periódicos o pseudoperiódicos generalizados de ondas agudas bifásicas o trifásicas de 1-2 ciclos/segundo que se observan con mayor frecuencia en las fases avanzadas de la enfermedad (11) y según Gao et al. se encuentran en el 63,5% de los casos (llegando al 90% en casos con mioclonías) (10). Se realizó un EEG a todos nuestros casos encontrándose los complejos típicos en el 80% de los casos probables y en el 50% de los definitivos (Figura 1), probablemente debido a que en los casos definitivos se realizó el EEG de forma más temprana.
El estudio del LCR muestra una discreta elevación del nivel de proteínas sin incremento de la celularidad (hallazgo no siempre presente) y se pueden encontrar bandas oligoclonales IgG. El análisis de algunas proteínas, como la proteína 14-3-3, es útil y su elevación en LCR representa daño neuronal aunque no es específico de ECJ (15) y tiene una mayor sensibilidad en la esECJ que en las otras formas. Según Chohan et al. la proteína 14-3-3 tiene una sensibilidad de 86% y una especificidad de 74% en fases avanzadas de la enfermedad (16). Se realizó la detección de la proteína 14-3-3 en tres de nuestros casos y solo en uno fue positivo. No se realizó en los demás casos por la reciente disponibilidad en nuestro país y alto costo de la prueba. Existen otras proteínas, como la proteína tau, la enolasa neuronal específica y el marcador astrocítico s100b que han mostrado una sensibilidad cercana a la proteína 14-3-3 y su combinación con esta muestra mayor especificidad (16).
En la mayoría de casos de esECJ la tomografía cerebral es normal o muestra diversos grados de atrofia cerebral. En la RM, las anomalías en los protocolos de difusión y FLAIR forman parte de los criterios diagnósticos actuales de ECJ probable. En esECJ, es característica la hiperintensidad bilateral del núcleo caudado, putamen, tálamo y áreas de la corteza cerebral, hallazgos presentes entre el 81 y el 91% de casos (14,17). Se realizó la RM con los protocolos mencionados en siete de nuestros casos y se observaron los hallazgos típicos en dos de los casos probables.
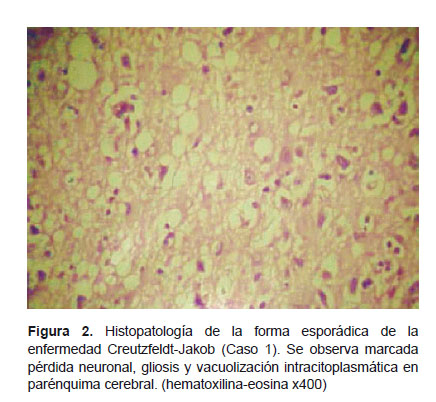
El diagnóstico definitivo de la ECJ solo puede ser realizado mediante el estudio histopatológico. Macroscópicamente, se observa distintos grados de atrofia cerebral y cerebelosa. En la microscopia de los pacientes con esECJ, tinciones como la hematoxilina-eosina, muestran la triada característica de vacuolización intracitoplasmática (cambio espongiforme), marcada pérdida neuronal y gliosis en zonas de la corteza cerebral, cerebelo y núcleos basales (18). Previa autorización del familiar directo, se realizó el estudio histopatológico post mortem a seis de nuestros casos y se observaron dichos cambios en todos nuestros casos definitivos (Figura 2). No se encontraron placas amiloides rodeadas de cambios espongiformes, típicas de la vECJ.
Existen reportes de esECJ en varios países de Sudamérica y se ha implementado un sistema de vigilancia de ECJ en países como Chile, Argentina, Uruguay y Brasil, con el objetivo de detectar de forma oportuna los casos de ECJ (en especial los de vECJ) debido a su riesgo potencial de transmisión. Actualmente la ECJ no forma parte de las enfermedades de notificación individual del sistema nacional de vigilancia epidemiológica del Perú por lo que sugerimos su inclusión.
En conclusión, se presentan seis casos de la forma esporádica de ECJ con diagnóstico definitivo por confirmación histopatológica, y cinco casos con diagnóstico probable, de ascendencia amerindia en Perú. Debe considerarse esta enfermedad en individuos mayores de 50 años, con una demencia rápidamente progresiva asociada a mioclonías, síntomas visuales y cerebelosos, con signos de disfunción piramidal y extrapiramidal. A pesar que el diagnóstico final es histopatológico, existen pruebas complementarias disponibles actualmente que ayudan a identificar con precisión y rapidez esta enfermedad.
Contribuciones de autoría: LTR, JRQ y CCE, participaron en la concepción, diseño, redacción y en la aprobación de su versión final. LTR, JRQ y DRF participaron en la recolección, análisis e interpretación de datos. LTR, JRQ, CCE, MVR, MFM, DRF, RSR y YNC participaron en la revisión crítica del artículo.
Fuentes de financiamiento: autofinanciado.
Conflictos de interés: los autores declaran no tener conflictos de interés.
Referencias Bibliográficas
1. Johnson RT. Prion diseases. Lancet Neurol. 2005;4(10):635-42. [ Links ]
2. Richardson EP Jr, Masters CL. The nosology of Creutzfeldt-Jakob disease and conditions related to the accumulation of PrPCJD in the nervous system. Brain Pathol. 1995;5(1):33-41. [ Links ]
3. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136-44. [ Links ]
4. Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11(7):618-28. doi: 10.1016/S1474-4422(12)70063-7. [ Links ]
5. Mead S, Stumpf MPH, Whitfield J, Beck JA, Poulter M, Campbell T, et al. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science. 2003;300(5619):640-3. [ Links ]
6. Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins S, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64(9):1586-91. [ Links ]
7. Safar JG. Molecular pathogenesis of sporadic prion diseases in man. Prion. 2012;6(2):108-15. doi: 10.4161/pri.18666. [ Links ]
8. Ironside JW. Variant Creutzfeldt-Jakob disease. Haemophilia. 2010;16 Suppl 5:175-80. doi: 10.1111/j.1365-2516.2010.02317.x. [ Links ]
9. Brown K, Mastrianni JA. The prion diseases. J Geriatr Psychiatry Neurol. 2010;23(4):277-98. doi: 10.1177/0891988710383576. [ Links ]
10. Gao C, Shi Q, Tian C, Chen C, Han J, Zhou W, et al. The epidemiological, clinical, and laboratory features of sporadic Creutzfeldt-Jakob disease patients in China: surveillance data from 2006 to 2010. PloS One. 2011;6(8):e24231. doi: 10.1371/journal.pone.0024231. [ Links ]
11. Wieser HG, Schindler K, Zumsteg D. EEG in Creutzfeldt-Jakob disease. Clin Neurophysiol. 2006;117(5):935-51. [ Links ]
12. Rubin M, Graff-Radford J, Boeve B, Josephs KA, Aksamit AJ. The alien limb phenomenon and Creutzfeldt–Jakob disease. Parkinsonism Relat Disord. 2012;18(7):842-6. doi: 10.1016/j.parkreldis.2012.04.009. [ Links ]
13. Head MW, Ironside JW. The contribution of different prion protein types and host polymorphisms to clinicopathological variations in Creutzfeldt-Jakob disease. Rev Med Virol. 2012;22(4):214-29. doi: 10.1002/rmv.725. [ Links ]
14. Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132(Pt 10):2659-68. doi: 10.1093/brain/awp191. [ Links ]
15. Stoeck K, Sanchez-Juan P, Gawinecka J, Green A, Ladogana A, Pocchiari M, et al. Cerebrospinal fluid biomarker supported diagnosis of Creutzfeldt-Jakob disease and rapid dementias: a longitudinal multicentre study over 10 years. Brain. 2012;135(Pt 10):3051-61. doi: 10.1093/brain/aws238. [ Links ]
16. Chohan G, Pennington C, Mackenzie JM, Andrews M, Everington D, Will RG, et al. The role of cerebrospinal fluid 14-3-3 and other proteins in the diagnosis of sporadic Creutzfeldt-Jakob disease in the UK: a 10-year review. J Neurol Neurosurg Psychiatry. 2010;81(11):1243-8. doi: 10.1136/jnnp.2009.197962. [ Links ]
17. Carswell C, Thompson A, Lukic A, Stevens J, Rudge P, Mead S, et al. MRI findings are often missed in the diagnosis of Creutzfeldt-Jakob disease. BMC Neurol. 2012;12:153. doi: 10.1186/1471-2377-12-153. [ Links ]
18. Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F, et al. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol. 1995;5(4):459-66. [ Links ]
Correspondencia: Luis Torres Ramírez
Dirección: Jr. Ancash 1271 Lima 1 Perú.
Teléfono: 4117700 – Anexo 232
Correo electrónico: torresramirezl@hotmail.com
Recibido: 29-11-13
Aprobado: 12-03-14
 Todo el contenido de esta revista, excepto dónde está identificado, está bajo una Licencia Creative Commons
Todo el contenido de esta revista, excepto dónde está identificado, está bajo una Licencia Creative Commons
Lima, Perú,
Apartado Postal 471
Teléfono : ( 51-1) 748-1111
Anexos 2122 / 2203

rpmesp@ins.gob.pe