










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
Print version ISSN 1726-4634
Rev. perú. med. exp. salud publica vol.31 no.4 Lima Oct./Dec. 2014
Reporte de Caso
Primer reporte de alcaptonuria en el Perú
First report of alkaptonuria in Peru
Daniel Guillén-Mendoza1,a, María Quiroga de Michelena1,2,b
1 Facultad de Medicina, Universidad Peruana Cayetano Heredia. Lima, Perú.
2 Genética. Instituto de Medicina Genética. Lima, Perú
a Médico Cirujano, b Médico Genetista, doctor en Medicina.
RESUMEN
La alcaptonuria es un error innato del metabolismo causado por la deficiencia de la homogentisiato 1,2 dioxidasa (HGD), produciéndose un exceso de ácido homogentísico (HGA). Se presenta el caso de una mujer de 57 años quien, desde que nació, su orina se tornaba de color negro; desde hacía 9 años presentaba una pigmentación verdosa en los lechos ungueales que no mejoró con tratamientos antifúngicos y en los últimos 9 meses presentó artrosis de articulaciones grandes que fue empeorando, forzándola a usar una silla de ruedas por el intenso dolor generado por la artrosis de caderas y columna lumbar. Por la descripción de los síntomas se le solicitó la medición de HGA en orina lo que confirmó el diagnóstico de alcaptonuria. Se sugirieron analgésicos, dieta sin productos que contuvieran tirosina y fue referida para cirugía de reemplazo de cadera. Se trata del primer reporte de caso de alcaptonuria en el Perú.
Palabras clave: Alcaptonuria; Metabolismo; Ácido homogentísico (fuente: DeCS BIREME).
ABSTRACT
Alkaptonuria is an inborn error of metabolism caused by deficiency of homogentisate 1,2-dioxygenase (HGD) which produces an excess of homogentisic acid (HGA). A case is presented of a 57 year old woman whose urine has turned black since birth. For 9 years she presented a greenish pigmentation in her nail beds that did not improve with antifungal treatments, and in the last 9 months she showed worsening large joint osteoarthritis. This situation forced her to use a wheelchair due to the intense pain caused by osteoarthritis in her hips and lumbar spine. From the description of symptoms, her urinary HGA was measured which confirmed the diagnosis of alkaptonuria. Analgesics and a diet without tyrosine-containing products were suggested. The patient was also referred for hip replacement surgery. This is the first reported case of alkaptonuria in Peru.
Key words: Alkaptonuria; Metabolism; Homogentisic acid (source: MeSH, NLM).
INTRODUCCIÓN
La alcaptonuria es el primer desorden metabólico autosómico recesivo, descrito por Garrod en 1902. Se debe a la deficiencia del la enzima HGD, por mutaciones del gen HGD en el cromosoma 3q21-q23 (1). Dicha enzima se encuentra en la vía de degradación de la tirosina (2) catalizando la conversión de HGA en maleilacetoacetato (3). Debido a la ausencia de HGD, el HGA se oxida, formando ácido benzoquino acético, el cual forma un polímero similar a melanina, responsable de la coloración negra en tejidos y orina (4). Esta enfermedad se caracteriza por una triada cronológica de signos: alcaptonuria, ocronosis y artropatía ocronótica. Hasta el momento se han reportado más de 700 casos de alcaptonuria en el mundo, siendo unas de las series más grandes las reportada por Al-Sbou, en Jordania, donde encontró 40 casos (5) y la revisión de casos de Aquaron, en Francia, con 96 casos y una prevalencia calculada para el país de 1:680 000 (1). En una revisión realizada en los buscadores de Pubmed, Lilacs y Scielo no se encontraron series ni reportes de casos de esta enfermedad en el Perú, este sería el primer caso reportado de esta enfermedad.
REPORTE DE CASOMujer de 57 años, natural de Sayán, en la provincia de Huaura, quien durante el último año antes del diagnóstico, presentó dolores articulares que iniciaron en la rodilla derecha y se fueron intensificando, se agregó, en los últimos 9 meses, dolor en la rodilla izquierda que empeoraban al caminar y le impedían la movilización, acudió a consulta de reumatología donde fue diagnosticada de artritis reumatoide (AR) y se le inició tratamiento con metrotexate; tratamiento que le generó malestar general y edemas en ambos pies; negó en todo momento rigidez matutina o dolor en manos. En los últimos dos meses empezó a presentar dolor en hombros, caderas y pies, lo cual la ha forzado a usar silla de ruedas; mantuvo el tratamiento para AR en forma irregular pero con empeoramiento del dolor, requirió múltiples analgésicos. Como antecedentes de importancia, la paciente nació sin recibir ninguna atención médica, en medio de una plantación, la madre se rehusó a darle de lactar, la alimentaba solo con café. Desde su nacimiento se observó que manchaba los pañales de color negro y que, al dejar reposar su orina, a pesar de haber sido de color claro, se tornaba oscura en poco tiempo; dicha molestia la ha presentado toda la vida y había consultado en múltiples ocasiones sin tener respuesta. No se conocen los antecedentes familiares de la paciente porque la madre la abandonó a los 15 días de vida y nunca se conoció al padre, aparentemente, no era quien la madre había declarado en el acta de nacimiento y existe la sospecha de consanguineidad de acuerdo con la información con la que cuentan los familiares.
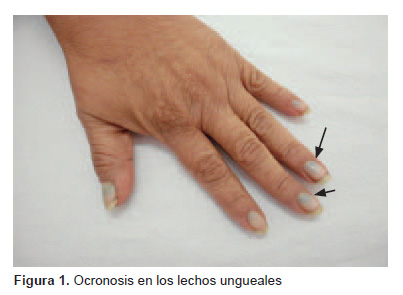
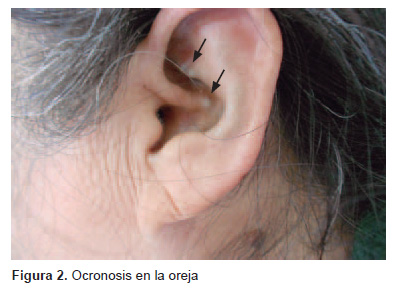
Desde hace 9 años presenta pigmentación verdosa en lechos ungueales (Figura 1). En el último año también presenta el mismo tipo de pigmentación en las orejas (Figura 2). La paciente consultó múltiples veces por ello y recibió varios cursos de tratamiento antifúngico sin ninguna mejora. Al examen físico, también se evidenció intenso dolor y crujido a la movilización de articulaciones grandes: rodillas, caderas, hombros y columna vertebral, en especial en la región lumbar, las articulaciones en las manos eran normales, no se observó deformación en forma de cuello de cisne ni rigidez; además, se observó edemas en ambos pies. Inicialmente se tomaron pruebas de laboratorio que documentaron VSG en 50 mm/h y PCR 3,2 mg/dL. El hemograma, las pruebas de función hepática y renal y el examen de orina fueron normales. Por el cambio de coloración de la orina se solicitó también uroporfirinas, que fueron normales, y por el diagnóstico previo de AR se solicitó factor reumatoideo, que fue negativo.
Se descartó la posibilidad de AR, por lo que se suspendió el metrotexate, con ello desapareció el edema y disminuyó el malestar general. Ante la sospecha de alcaptonuria se solicitó HGA en orina, el cual fue positivo, y se confirmó el diagnóstico. Se inició tratamiento a la paciente con múltiples analgésicos y dieta estricta evitando alimentos que contengan tirosina. Actualmente, la artrosis ha empeorado, en especial en las caderas, por lo que no ha vuelto a caminar.
DISCUSIÓNLa alcaptonuria es una rara enfermedad hereditaria del catabolismo de la tirosina, autosómico recesiva, caracterizada por la presencia de ácido homogentísico en orina. Su prevalencia aproximada es de 1:100 000 a 1:1 000 000 de habitantes (1,6). Estos pacientes, tienen deficiencia de HGD, lo que origina depósitos de HGA en diversos tejidos del organismo ricos en colágeno (ocronosis). Se han identificado hasta 16 tipos de mutaciones en el gen HGD humano (cromosoma 3), que pueden provocar una pérdida de función de la enzima (7). También hay una posible asociación con la presencia de HLA B-27 (8). La alcaptonuria da lugar a la aparición de una orina de color negro-pardusca, como consecuencia de la oxidación del ácido homogentísico que contiene (6). Los pacientes alcaptonúricos que viven hasta la quinta década de la vida, casi invariablemente presentan ocronosis (9).
Los pacientes ocronóticos desarrollan espondilosis ocronótica, caracterizada por el envaramiento progresivo de la columna lumbar y pérdida de la lordosis fisiológica, casi siempre de forma sintomática. En el caso de nuestra paciente, este cuadro se ve reflejado en el intenso dolor lumbar que presentaba. En una gran proporción de hombres con ocronosis aparecen cálculos prostáticos de color negro (10). En referencia a otras zonas de depósito del ácido homogentísico, se sabe que existen afectaciones cardiovasculares de alcaptonuria, con depósitos y posterior lesión en válvulas cardiacas, pericardio y arterias coronarias (11). También existen reportes de ruptura de tendones no traumática (12,13).
El diagnóstico de la enfermedad se realiza a través de la detección del HGA en orina, utilizando una prueba muy sencilla y de bajo costo que, de ser aplicada a paciente con síntomas compatibles, permitiría el diagnóstico correcto en algunas de ellas. Pero para ello es necesario que el médico conozca los síntomas y sospeche la enfermedad. Con respecto al tratamiento, inicialmente se recomienda evitar el consumo de alimentos que contengan tirosina, sin embargo, en nuestra paciente, los depósitos de HGA ya están formados y no se espera que esta dieta tenga un gran impacto. Por otro lado, para el manejo de la artrosis severa que presenta se vienen ensayando cirugías de reemplazo de cadera (14), lo cual sería lo recomendado para nuestra paciente. Recientemente se ha desarrollado un tratamiento potencial con un medicamento llamado nitisinona, que disminuiría la formación de HGA, evitando la unión a articulaciones. Estudios en modelos animales han demostrado buena eficacia del tratamiento (15); sin embargo, es necesario conocer adecuadamente la historia natural de la enfermedad, por lo cual se vienen haciendo estudios de cohorte que permitan conocer mejor la evolución (9).
Presentamos el primer reporte de caso de alcaptonuria diagnosticado en Perú, con evidencia de la triada clásica descrita por Garrod de alcaptonuria, ocronosis y artropatía ocronótica.
Contribuciones de autoría: DGM y MQM han participado en la concepción del artículo, la recolección de datos, su redacción y aprobación de la versión final.
Referencias Bibliográficas
1. Aquaron RR. Alkaptonuria in France: past experience and lessons for the future. J Inherit Metab Dis. 2011 Dec;34(6):1115-26. doi: 10.1007/ s10545-011-9392-7 [ Links ]
2. Fernández-Cañón JM, Granadino B, Beltrán-Valero de Bernabé D, Renedo M, Fernández-Ruiz E, Peñalva MA, et al. The molecular basis of alkaptonuria . Nat Genet. 1996 Sep;14(1):19-24. [ Links ]
3. La Du BN, Zannoni VG, Laster L, Seegmiller JE. The nature of the defect in tyrosine metabolism in alcaptonuria . J Biol Chem. 1958;230(1):251-60. [ Links ]
4. Zannoni VG, Lomtevas N, Goldfinger S. Oxidation of homogentisic acid to ochronotic pigment in connective tissue . Biochim Biophys Acta. 1969 Feb 18;177(1):94-105. [ Links ]
5. Al-Sbou M, Mwafi N, Lubad MA. Identification of forty cases with alkaptonuria in one village in Jordan . Rheumatol Int. 2012 Dec;32(12):3737-40. doi: 10.1007/ s00296-011-2219-x. [ Links ]
6. Aquaron R. Alkaptonuria: a very rare metabolic disorder . Indian J Biochem Biophys. 2013 Oct;50(5):339-44. [ Links ]
7. Zatkova A, Sedlackova T, Radvansky J, Polakova H, Nemethova M, Aquaron R, et al. Identification of 11 Novel Homogentisate 1,2 Dioxygenase Variants in Alkaptonuria Patients and Establishment of a Novel LOVD- Based HGD Mutation Database . JIMD Rep. 2012;4:55-65. doi: 10.1007/8904_2011_68. [ Links ]
8. John SS, Padhan P, Mathews JV, David S. Acute anterior uveitis as the initial presentation of alkaptonuria . J Postgrad Med. 2009 Jan-Mar;55(1):35-7. [ Links ]
9. Ranganath LR, Cox TF. Natural history of alkaptonuria revisited: analyses based on scoring systems . J Inherit Metab Dis. 2011 Dec;34(6):1141-51. doi: 10.1007/s10545-011-9374-9. [ Links ]
10. Scriver CR. Garrods Croonian Lectures (1908) and the charter Inborn Errors of Metabolism: albinism, alkaptonuria, cystinuria, and pentosuria at age 100 in 2008 . J Inherit Metab Dis. 2008 Oct;31(5):580-98. doi: 10.1007/s10545-008-0984-9. [ Links ]
11. Pettit SJ, Fisher M, Gallagher JA, Ranganath LR. Cardiovascular manifestations of Alkaptonuria . J Inherit Metab Dis. 2011 Dec;34(6):1177-81. doi: 10.1007/s10545-011-9339-z. [ Links ]
12. Chua SY, Chang HC. Bilateral spontaneous rupture of the quadriceps tendon as an initial presentation of alkaptonuria--a case report. Knee. 2006 Oct;13(5):408-10. [ Links ]
13. Logani V, Eachempati KK, Malhotra R, Bhan S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2004 Sep;86(7):1090. [ Links ]
14. Kerimoglu S, Onder C, Aynaci O, Malkoc CH. Hip arthroplasty for ochronosis. Saudi Med J. 2005 Nov;26(11):1812-4. [ Links ]
15. Preston AJ, Keenan CM, Sutherland H, Wilson PJ, Wlodarski B, Taylor AM, et al. Ochronotic osteoarthropathy in a mouse model of alkaptonuria, and its inhibition by nitisinone. Ann Rheum Dis. 2014 Jan;73(1):284-9. doi: 10.1136/annrheumdis-2012-202878. [ Links ]
Correspondencia: Daniel Guillén Mendoza. Dirección: 245 Ramón Castilla, Ingeniería, Lima 31, Perú Teléfono: 996550544 Correo electrónico: daniel.guillen@upch.pe
Recibido: 20-05-14
Aprobado: 15-10-14

