










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
versión impresa ISSN 1726-4634
Rev. perú. med. exp. salud publica vol.32 no.4 Lima oct. 2015
Artículo de Revisión
La obesidad como factor de riesgo en el desarrollo de cáncer
Obesity as a risk factor in the development of cancer
Deissy Herrera-Covarrubias1,a, Genaro Alfonso Coria-Avila1,b, Cynthia Fernández-Pomares1,c, Gonzalo E. Aranda-Abreu1,d, Jorge Manzo Denes1,e, María Elena Hernández1,f
1 Centro de Investigaciones Cerebrales. Universidad Veracruzana. Veracruz, México.
a Licenciada en Nutrición, doctora en Neuroetología; b médico veterinario zootecnista, doctor en Psicología Experimental; c química farmacéutica bióloga, doctora en Neuroetología; d licenciado en Biología, doctor en Neurobiología; e licenciado en Biología, doctor en Ciencias Fisiológicas; f licenciada en Biología, doctora en Ciencias Fisiológicas, investigador titular.
RESUMEN
De acuerdo con la Organización Mundial de la Salud, cada año fallecen 3,4 millones de personas adultas por consecuencias del sobrepeso u obesidad. Personas con un índice de masa corporal superior a 30, presentan cierto aumento en la incidencia de algunas enfermedades entre las que se encuentran algunos tipos de cánceres. En esta revisión de tipo narrativa se aborda el papel que tiene el tejido adiposo como modulador del sistema endocrino y facilitador de la inflamación crónica subclínica. Se discute cómo la obesidad puede producir un microambiente favorable para el desarrollo de tumores, principalmente por el incremento del estrés oxidativo y en las concentraciones de diversas hormonas como la leptina, la insulina y la prolactina. Se concluye que, en conjunto, estos factores incrementan la probabilidad de desarrollar cáncer.
Palabras clave: Obesidad; Cáncer; Hormonas; Inflamación; Estrés oxidativo (fuente: DECS/Bireme)
ABSTRACT
According to the World Health Organization, every year about 3.4 million adults die of consequences related to overweight or obesity. People with a Body Mass Index above 30 are more likely to express certain diseases, including some types of cancer. In this narrative review, we assess the role of adipose tissue as a modulator of the endocrine system and facilitator of chronic subclinical inflammation. We discuss how obesity can induce a suitable micro environment for the development of tumors, mainly by enhancing the levels of oxidative stress and the concentrations of hormones such as leptin, insulin and prolactin. We conclude that all together, these factors increase the probability of cancer development.
Key words: Obesity; Cancer; Hormones; Inflammation; Oxidative stress (source: MeSH/NLM)
INTRODUCCIÓN
En la actualidad, la obesidad es considerada dentro del grupo de enfermedades crónicas que afectan a la población infantil y adulta de muchos países. De acuerdo con la Organización Mundial de la Salud (OMS) la obesidad es definida como una acumulación anormal excesiva de grasa perjudicial para la salud. El exceso de grasa se calcula, generalmente, en base al índice de masa corporal (IMC) considerando el peso y la talla de una persona, de tal forma que un IMC normal oscila entre 18,5–24,9, mientras que un IMC ≥ 25 indica sobrepeso, ≥ 30 obesidad, y ≥ 40 obesidad mórbida (1). Se cree que entre mayor sea el IMC, mayor es la probabilidad de desarrollar enfermedades. Lo anterior es debido a que el sobrepeso y la obesidad resultan en mayores niveles de estrés oxidativo que puede producir inflamación crónica subclínica, así como alteraciones en el funcionamiento del sistema inmune. Además, las células del tejido adiposo no solo almacenan lípidos en el citoplasma para ocuparse como fuente de energía, sino que también pueden producir algunos tipos de hormonas. Por lo tanto, el aumento del tejido adiposo que ocurre en la obesidad crónica puede llevar a diferentes enfermedades que van desde alteraciones en el metabolismo, hasta algunos tipos de neoplasias desencadenadas por hormonas. En esta revisión de tipo narrativa se discuten datos experimentales y clínicos que muestran el papel de la obesidad como un factor muy importante en el desarrollo de diferentes tipos de cáncer.
ESTADÍSTICAS DE LA OBESIDAD EN AMÉRICA LATINA
Un reporte de la OMS en 2014 indicó que a nivel mundial existían 1900 millones de adultos (mayores de 18 años) con sobrepeso y 600 millones con obesidad, siendo más frecuente el sobrepeso y obesidad en mujeres que en hombres (2). La Organización de Naciones Unidas para la Alimentación y la Agricultura (FAO) reportó que: La prevalencia global de sobrepeso y obesidad ha aumentado en todas las regiones y también está aumentando en casi todos los países con una prevalencia de más del 30%, encabezando la lista los países de Latinoamérica y el Caribe (3). A nivel internacional ya se considera el sexto factor principal de riesgo de defunción, pues cada año mueren 3,4 millones de personas a causa de sus complicaciones. Así mismo, se ha calculado que para el año 2020, países latinoamericanos ocuparán los primeros seis lugares en padecimientos relacionados con sobrepeso y obesidad (4).
LA OBESIDAD COMO FACTOR DE RIESGO EN EL CÁNCER
La OMS define al cáncer como “un proceso incontrolado del crecimiento y diseminación de células que puede aparecer en cualquier lugar del cuerpo” (5). Un tumor formado por células cancerosas suele invadir el tejido circundante y también puede provocar metástasis en puntos distantes del organismo. Debido a las alteraciones funcionales que produce en órganos vitales, el cáncer es una de las principales causas de mortalidad a nivel mundial. Tan solo en 2012 se detectaron 14 millones de casos, de los cuales 8,2 millones (>50%) resultaron en defunciones. Se ha estimado que en aproximadamente dos décadas la incidencia será de 22 millones de casos, lo cual pudiera resultar en más de 10 millones de muertes anuales (6).
Existen diversos factores de riesgo en el desarrollo del cáncer. Los más conocidos son la edad, la etnia, el sexo, la genética, la exposición a sustancias químicas, la radiación y el tabaquismo (7). Recientemente se ha comenzado a considerar a la obesidad como un factor de riesgo más, pues se correlaciona con una mayor probabilidad de desarrollar algún tipo de cáncer. Desafortunadamente, las causas exactas que vinculan a estos dos padecimientos de salud pública no están del todo esclarecidas, pero algunos investigadores han llegado a sugerir que entre el 35- 70% de los cánceres están vinculados directamente con la alimentación (8,9) y con un IMC elevado que refleja obesidad (7). Por ejemplo, en 1984 Snowdon et al. publicaron un estudio de casos y controles que había comenzado dos décadas atrás con 6763 hombres a los cuales se les evaluó la dieta, el peso y la causa de defunción (10). Los hallazgos indicaron que aquellos con sobrepeso tuvieron 2,5 veces más probabilidad de morir de cáncer prostático. Dicha probabilidad aumentaba a 3,6 veces si el sobrepeso se asociaba al consumo de grasas de origen animal (leche, queso, huevos y carne). En la Tabla 1 se indican las posibles consecuencias en la salud de acuerdo al incremento de IMC. En los casos más severos la obesidad se asocia con el desarrollo de diversos tipos de cáncer (7).
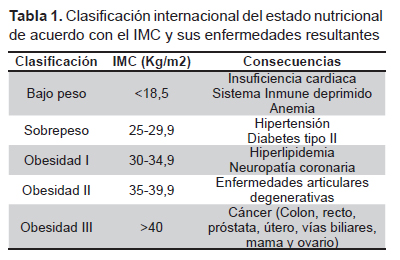
Cabe mencionar que cuando otros factores de riesgo se suman a la obesidad, la probabilidad de desarrollar cáncer es mucho mayor. Por ejemplo, en cuanto al género, puede resultar en mayor incidencia de cáncer colorrectal en hombres que en mujeres, diferencia que puede ser atribuible a hormonas como los andrógenos (11). Además, estudios han mostrado que las personas con un IMC ³25 presentan un mayor riesgo de presentar de este tipo de cáncer (12).
Así mismo, la obesidad y el género resultan en mayor probabilidad de cáncer de mama en mujeres con sobrepeso, diferencia relacionada a otras hormonas como los estrógenos (13,14). Estudios clínicos también han mostrado que mujeres con obesidad mórbida (IMC ≥ 40) presentan cáncer de mama a edades más tempranas (~32 años) que aquellas mujeres con antecedentes familiares de cáncer, pero que mantenían un peso normal (55 años aproximadamente) (15). Otro estudio realizado por Haakinson et al. mostraron que mujeres obesas presentaban tumores más grandes y eran más propensas a desarrollar metástasis en nódulos linfáticos. Los autores también reportaron que mujeres obesas presentaban cáncer de mama en etapa más avanzada comparadas con mujeres no obesas, probablemente porque la obesidad también pudiera facilitar la presencia de tumores no palpables (16). Además, existe otro factor de riesgo implicado en el cáncer de mama, como lo es la densidad mamaria, dependiente, en parte, de la cantidad de tejido graso en la glándula. Se ha reportado que mujeres con alta densidad mamaria tienen de cuatro a seis veces mayor riesgo de cáncer (17,18).
La obesidad también es un riesgo para desarrollar otros tipos de cáncer como de endometrio y de ovario, los cuales están fuertemente asociados con cambios hormonales (19). Se ha considerado que el tejido adiposo puede funcionar como un órgano con funciones endocrinas múltiples (20), pero aún se desconoce todo su potencial como modulador del sistema endocrino. Dicho potencial puede alterar el metabolismo celular e incrementar la proliferación celular de manera anormal. Entre otras posibles explicaciones hormonales que vinculan a la obesidad y el cáncer se incluyen las alteraciones en los niveles de leptina, insulina, prolactina, cortisol y andrógenos (14,21-23).
CONSECUENCIAS ENDOCRINAS DE LA OBESIDAD Y SU IMPLICACIÓN EN EL CÁNCER
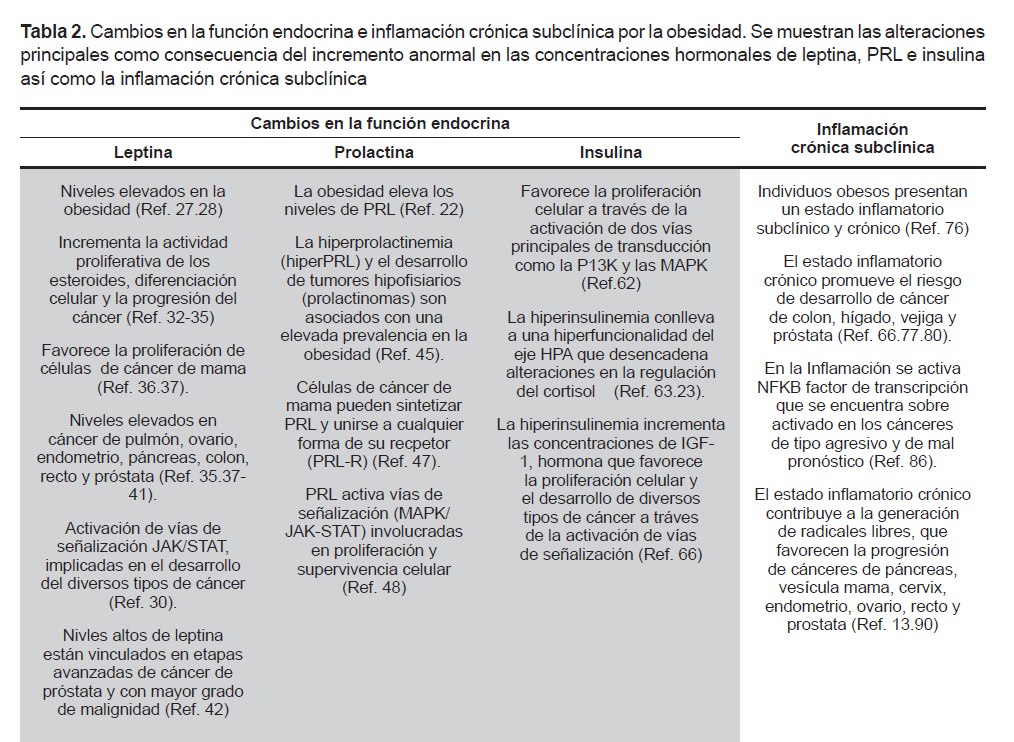
Las hormonas leptina, prolactina e insulina participan en la regulación del apetito, del metabolismo, del sistema inmune y cumplen una función fundamental en la génesis tumoral. Por ello, las alteraciones en sus niveles han sido asociadas como un factor de riesgo para desarrollar algún tipo de cáncer (24-26). Su importancia radica en que participan en la cascada de señalización de las proteínas transductoras de señales y activadoras de la transcripción (STAT), la proteína cinasa activada por mitógenos (MAPK) y la fosfatidilinositol 3-cinasa (PK13), vías que regulan la expresión génica y mitogénica (Tabla 2).
LEPTINA
La leptina es secretada primordialmente por los adipocitos de la grasa subcutánea, y la cantidad liberada es directamente proporcional a la masa del tejido adiposo (27,28). Su receptor (OBR) pertenece a la gran familia de receptores de citocinas de clase I y se expresa en diversos órganos como la placenta, endometrio, ovario, estómago, páncreas, colon, pulmón e hipotálamo (29). La función principal de la leptina es la regulación del apetito, el balance energético y el metabolismo de las grasas y la glucosa (30).
Normalmente su incremento inhibe el apetito a través de la vía hipotalámica, actuando sobre el núcleo arcuato, ventromedial y dorsomedial, donde desencadena una cascada de señales específicas que inhiben diversos péptidos orexigénicos (21,31). Sin embargo, algunos estudios han encontrado que la leptina puede incrementar la actividad proliferativa de los esteroides, la diferenciación celular y la progresión del cáncer (32- 35). En ensayos in vitro e in vivo se ha observado que puede favorecer la proliferación de células de carcinoma de mama (36,37). Además, sus niveles también están incrementados en casos de cáncer de pulmón, ovario, endometrio, páncreas, colon, recto y próstata (35,37-41).
El desarrollo del cáncer de próstata también ha sido vinculado con la obesidad y niveles altos de leptina y sus receptores OBR (42). Un estudio en Suecia con 149 hombres diagnosticados con cáncer de próstata e IMC>25 mostró la presencia de receptores OBR en etapas avanzadas del desarrollo del tumor y en los mayores grados de malignidad (42). Con lo anterior se correlaciona que la obesidad parece estar ligada no solo al cáncer per se sino a las lesiones de alto grado de malignidad, lo cual da un pronóstico menos favorecedor para el paciente. La activación del receptor OBR puede desencadenar el inicio de vías de señalización a través de la tirosina cinasa (JAK2) (30), cuya activación lleva a la fosforilación de las STAT (43). Estas últimas son proteínas primordiales en las respuestas inmunes y en el microambiente tumoral donde pueden promover o inhibir el cáncer (44). Las STAT, a su vez, activan la transcripción de genes implicados en la diferenciación y proliferación celular. La vía JAK-STAT es uno de los principales mecanismos utilizados por una variada cantidad de citocinas y parece ser fundamental como vía de señalización para citocinas de clase I, como son: prolactina, hormona del crecimiento (GH) y la leptina. Además, la JAK2 también puede activar otras vías de señalización intracelulares como la MAPK, y la P13K (30,43), vías implicadas en el desarrollo de diferentes tipos de cáncer. De este modo, la leptina posee una vía metabólica que funciona como facilitadora de la proliferación celular, lo cual puede favorecer el desarrollo de tumores.
PROLACTINA
Otra hormona que se encuentra vinculada con la obesidad es la prolactina (PRL). Por ejemplo, se ha reportado que la hiperprolactinemia (hiperPRL) puede estar asociada con una elevada prevalencia en la obesidad. En dicho estudio se exploró la relación entre la hiperPRL y el peso corporal de pacientes con tumores hipofisiarios como el prolactinoma. Se reportó que la obesidad en pacientes con este tipo de tumores se presenta de manera independiente a las consecuencias que el tumor ejerza sobre el hipotálamo o la hipófisis.
Además, el 70% de los pacientes disminuyeron su peso corporal cuando los niveles de PRL se normalizaron (45). Una explicación del por qué la hiperPRL está asociada con la obesidad, puede deberse a que un incremento en los niveles de PRL induce la expresión del neuropéptido Y (NPY) en el núcleo hipotalámico dorsomedial, lo que conlleva a la hiperfagia (46). Por lo tanto, individuos con niveles altos de PRL tienden a consumir más alimento. Cabe mencionar que la misma obesidad parece también incrementar los niveles de PRL. Por ejemplo, en un estudio se reportó que ratones de laboratorio obesos tenían niveles de PRL en sangre hasta un 300% por arriba de los ratones no obesos. Se mostró que la restricción calórica que los hacía reducir el peso corporal retornaba los niveles de PRL a un nivel normal. Si esos mismos ratones consumían alimento ad libitum, su peso volvía a incrementar junto con niveles anormales de PRL (22).
Por otra parte, los niveles elevados de PRL también tienen un papel importante en el desarrollo de cáncer que se ha discutido ampliamente con evidencia in vitro e in vivo. Por ejemplo, Mujagić et al. (47) mencionan que la PRL puede sintetizarse por las mismas células de cultivo de cáncer de mama y puede unirse a cualquier isoforma de su receptor (PRLR) presente en el epitelio de la glándula mamaria. El complejo PRL/ PRLR activa diversas vías de señalización celular que se comparten con otros receptores de la superfamilia de las citocinas, como la leptina. Se ha sugerido que una vía de activación del PRLR es a través de las MAPK (48). Las cuales se encuentran involucradas en acciones proliferativas y de supervivencia celular. Por lo tanto, la activación constante y desmesurada de esta vía promueve la proliferación celular, lo que conlleva al crecimiento del tumor e incluso su invasión. La PRL también puede actuar como un factor de progresión. Es decir, un facilitador de alteraciones histológicas no cancerosas a cancerosas. Esto pudiera ocurrir, en parte, por su efecto como promotor del crecimiento tisular.
En pacientes con cáncer prostático, los niveles de prolactina en sangre son mayores al compararse con personas de la misma edad sanas (49,50). Si bien esta glándula depende de concentraciones fisiológicas normales de PRL, puede producirla en su epitelio secretor (51) o recibirla del tejido adiposo (52-54), lo cual se incrementa significativamente con la obesidad (55). El aumento en los niveles de PRL en sangre parecen influenciar, principalmente, las primeras etapas del desarrollo de cáncer prostático. Paradójicamente un estudio mostró que cuando el cáncer tiene grados avanzados de malignidad parece depender menos de la PRL, pues la expresión del receptor disminuye en próstatas con displasia de alto grado (4 o más en Gleason) (56). Sin embargo, se ha discutido que los carcinomas con alto grado de malignidad pudieran tener células pobremente diferenciadas al tejido y, por lo tanto, expresan menos receptores (57).
Hasta ahora los reportes omiten por completo los datos sobre los grados de sobrepeso u obesidad de los pacientes. Por lo tanto, desconocemos si la expresión del receptor a prolactina en la próstata pudiera depender de los niveles de prolactina asociados con la obesidad y no solo por el grado de malignidad de la lesión. Aunado a lo anterior, un estudio realizado en 2003, con ratones transgénicos con adenocarcinoma de próstata, mostró que la severidad de las lesiones en la próstata eran mucho menores en ratones que habían tenido una restricción calórica en la dieta, mientras que los alimentados de manera ad libitum tenían peor pronóstico. La conclusión del estudio sugirió que la reducción en la cantidad de grasa corporal podía beneficiar a los ratones, incluso si habían sido genéticamente manipulados para desarrollar cáncer de próstata (58). Adicionalmente, se ha mostrado que la PRL induce hiperinsulinemia y resistencia a la insulina (59). Esta última es considerada como factor de riesgo para el síndrome metabólico. Algunos estudios indican que la PRL puede ser un factor predictivo en la progresión del síndrome metabólico (60), el cual favorece la incidencia y la mortalidad en pacientes con carcinoma hepatocelular (61).
INSULINA
La hiperinsulinemia es el cambio endocrino más frecuentemente observado en la obesidad. La insulina es un hormona involucrada en procesos de proliferación celular a través de la activación de dos vías principales de transducción como la P13K y las MAPK (62). La hiperinsulinemia crónica, eventualmente, puede llevar a la resistencia a las acciones de la misma hormona. Se ha reportado que la resistencia de esta hormona está correlacionada con la obesidad de tipo central o también denominada androide, la cual también ha sido asociada a una hiperfuncionalidad del eje hipotálamo – pituitaria – adrenal (HPA) (63). Una hiperfuncionalidad de este eje puede desencadenar perturbaciones en la regulación de otras hormonas como el cortisol (23), el cual puede afectar el metabolismo de carbohidratos y lípidos que favorecen la acumulación abdominal de grasa. Esto pudiera deberse a que el cortisol en presencia de insulina produce una acumulación de lípidos, por lo tanto, este desbalance puede estar correlacionado con acumulación de grasa visceral, además de otros factores (64). Cabe mencionar que el cortisol es considerado uno de los desencadenantes de la resistencia a la insulina. El tejido adiposo responde de manera eficaz a los efectos de la insulina, la cual promueve la captación de triglicéridos. Por lo tanto, en los individuos obesos, los ácidos grasos libres están incrementados, con lo cual disminuye de manera importante la utilización de glucosa como fuente de energía. Esto conduce a una disminución en su captación, desencadenando su resistencia. El incremento de ácidos grasos origina la activación de diversas vías metabólicas que provocan alteraciones en el metabolismo celular (65).
La hiperinsulinemia también desencadena un incremento en las concentraciones del factor de crecimiento parecido a la insulina (IGF-1) (66). Niveles altos de IGF-1 son determinantes en la proliferación y apoptosis en el cáncer a través de la vía de señalización de la MAPK (66) y se han asociado con mayor riesgo de cáncer de próstata, mama, colon y pulmón (67-70). Así mismo, la insulina y el IGF-1 tienen efecto sobre las hormonas esteroideas que regulan el crecimiento celular y diferenciación en la mama, útero y otros tejidos como la próstata (30). De hecho, otra hormona esteroidea como la dihidrotestosterona (DHT) que se conoce por su efecto en las alteraciones prostáticas, también modula la expresión de IGF-I, IGFBP-2 y IGFBP-3 en células cancerígenas estromales prostáticas (71). Un estudio realizado por Baquedano et al., (72) reportó que el eje GH - IGF-1 es determinante en la acumulación de tejido adiposo y la respuesta inflamatoria a la obesidad inducida por una dieta alta en grasas. Estudios han vinculado a la GH y el IGF-1 con el desarrollo de tumores, esto debido a que poseen actividad mitogénica y antiapoptótica (73). Se sabe que la cascada de señalización producida por insulina/IGF posee un papel en el desarrollo y progresión de diversos tipos de cáncer. Así mismo, estas hormonas junto con la inflamación crónica de bajo grado son factores importantes y determinantes en promoción del cáncer inducido por la obesidad (74).
INFLAMACIÓN CRÓNICA INDUCIDA POR LA OBESIDAD Y SU REPERCUSIÓN EN EL CÁNCER
Se ha vinculado a la obesidad con un estado inflamatorio subclínico y crónico, el cual se presenta como consecuencia del incremento de tejido adiposo. Una posible explicación obedece a que el tejido adiposo secreta citocinas tales como el factor de necrosis tumoral (TNFα), y diversas interleucinas (IL-1, IL-6, IL-18) y quimiocinas. Estas últimas son un grupo de proteínas de bajo peso molecular que son secretadas también por células del sistema inmune (linfocitos, monocitos y macrófagos) y actúan en procesos inflamatorios (1,75). Por lo tanto, es de esperar que las personas obesas presenten con mayor frecuencia un estado inflamatorio crónico en un bajo grado (76), el cual también ha sido asociado como un factor de riesgo del desarrollo de cáncer. Al respecto, existen reportes que señalan que los procesos inflamatorios crónicos aumentan el riesgo de desarrollar cáncer de colon, hígado, vejiga y próstata (67,77-80). El estado inflamatorio crónico promueve los procesos carcinogénicos, estimula el crecimiento de las células tumorales y facilita la inestabilidad de su genoma, favoreciendo así la progresión y la diseminación del tumor.
La secreción de moléculas proinflamatorias por los adipocitos favorece la proliferación celular y atrae células del sistema inmune como macrófagos y linfocitos que, a su vez, también secretan estas moléculas inflamatorias. Así, el tejido adiposo en las personas obesas se encuentra con una elevada infiltración de macrófagos que poseen un papel primordial en las vías inflamatorias asociadas a la obesidad (81). Además, estas células alteran la diferenciación, la proliferación y el metabolismo de las células adiposas, y su acumulación también ha sido asociada con la resistencia a la insulina (82). El TNFα producido por los linfocitos, macrófagos y adipocitos puede inducir resistencia a la insulina, al igual que la IL-6 (83). En el proceso inflamatorio, el TNFα favorece el incremento en la concentración de triglicéridos a través de la producción de lipoproteínas de muy baja densidad (VLDL) (84). Por otra parte la IL-6, es una citocina implicada en la inflamación que participa en reacciones locales y sistémicas (1). La concentración de IL-6 en plasma parece ser proporcional a la grasa corporal, por lo tanto, el tejido graso es fuente primordial para esta citocina y se ha reportado que existe una correlación entre los niveles séricos de IL-6 y el nivel de obesidad que se presenta (85). El receptor de IL-6 es estructuralmente similar al receptor OBR, donde ambos activan la vía JAK/STAT que, como se ha mencionado, está vinculada con diferentes tipos de cánceres (1,44). El TNFα, la IL-6 y otras citocinas proinflamatorias promueven la activación de el factor nuclear Kappa B (NFκB), que es un factor de transcripción que regula genes implicados en inflamación, estrés, proliferación y la supervivencia celular, entre otros procesos. En células cancerosas a menudo se encuentra sobreactivado y su alta expresión está correlacionada con los cánceres de tipo agresivo y de mal pronóstico (86). En células no estimuladas el NF-κB se encuentra inactivo en el citosol asociado a proteínas inhibitorias (87,88); sin embargo, diversos factores como las radiaciones ultravioleta, citocinas, TNFα, interleucinas, especies reactivas de oxígeno (ERO) y radicales libres (89) favorecen la activación de NF-κB y su translocación al núcleo, desencadenando todas sus funciones proinflamatorias a través de la secreción de moléculas como citocinas y quimiocinas que, a su vez, atraen a células del sistema inmune, sosteniendo así el estado inflamatorio crónico.
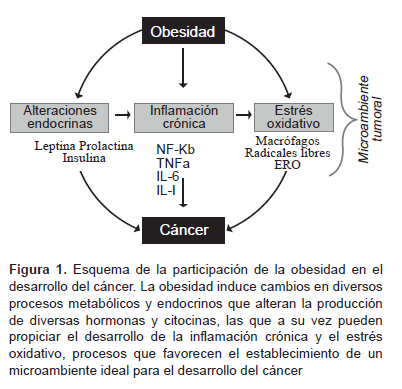
El estado inflamatorio crónico, marcado por la alta producción de citocinas, contribuye en gran medida a la generación de radicales libres. Algunos estudios indican una relación estrecha entre el estrés oxidativo y el desarrollo de diversos tipos de cánceres, entre ellos el de páncreas, vesícula, mama, cérvix, endometrio, ovario, recto y próstata (13,90). De manera general se considera que el desbalance que existe cuando el número de radicales libres excede el de moléculas antioxidantes se conoce como estrés oxidativo (91), y como resultado el ADN se daña dando origen a la aparición de mutaciones, inestabilidad genómica de la célula y al consecuente desarrollo del proceso maligno (92). En la obesidad, por lo general, se presenta un estado alterado de hiperglucemia que puede desencadenar un estrés oxidativo debido a la oxidación de la glucosa, favoreciendo con esto a la formación de ERO (93). La acumulación de grasa está correlacionada con el estrés oxidativo sistémico tanto en humanos y en modelos animales (94). Esto indica que el estrés oxidativo es un catalizador para las enfermedades asociadas a la obesidad. Las ERO y los radicales libres han sido vinculados en el desarrollo de diversos tipos de cáncer, pues son moléculas altamente reactivas capaces de dañar a proteínas, lípidos y el ADN de la célula a través de mecanismos de oxidación. En la Figura 1 se muestra la estrecha relación entre la obesidad y sus consecuencias endocrinas y metabólicas, las cuales, en conjunto, favorecen el proceso proliferativo o inflamatorio que conlleva a un elevado estrés oxidativo.
METFORMINA COMO AGENTE ANTIOBESIDAD Y SU EFECTO PROTECTOR EN EL CÁNCER
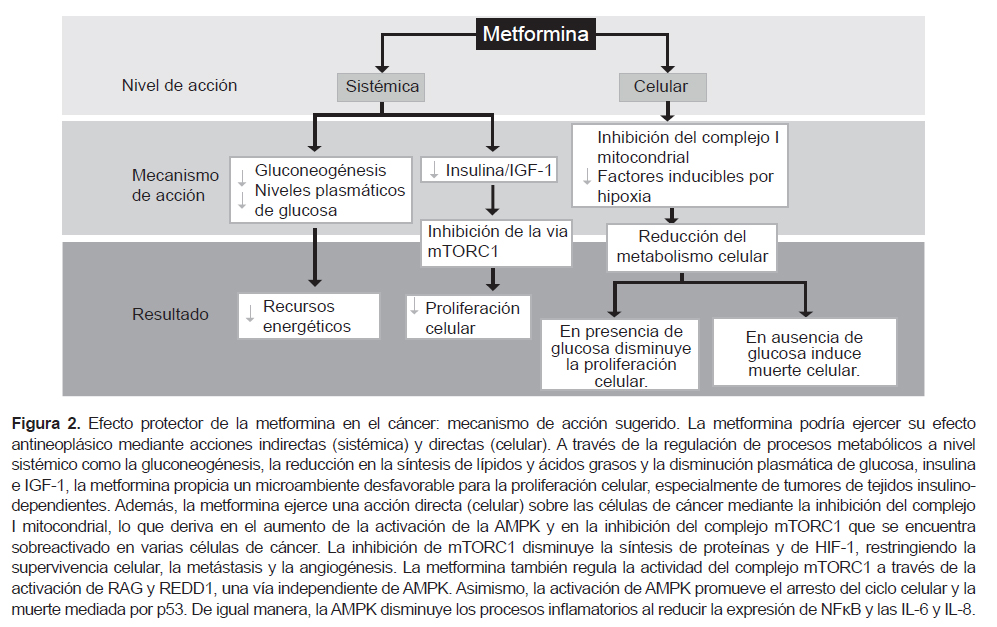
La metformina es uno de los fármacos de primera elección en el tratamiento de la diabetes mellitus tipo 2 y es empleada en pacientes con síndrome de ovario poliquístico. Estudios clínicos y preclínicos han señalado el potencial de esta biguanida como agente antineoplásico, planteado dos posibles vías de acción: una vía indirecta a través de su efecto antidiabético a nivel sistémico y una vía directa sobre las células de cáncer; sin embargo, aún permanece por elucidar el mecanismo a través del cual pudiera ejercer sus efectos contra el cáncer. Ambos mecanismos de acción no son excluyentes, por el contrario podrían compartir algunos procesos moleculares (95) (Figura 2).
Las células tumorales se caracterizan por tener un metabolismo alterado ya que consumen altos niveles de glucosa (96). La diabetes mellitus tipo 2 aumenta el riesgo de desarrollar varios tipos de cáncer, como de páncreas, hígado, mama, colorrectal y ginecológicos. El aumento de los niveles circulantes de glucosa y de los agentes mitógenos, insulina y IGF-1, favorecen la activación de las vías de señalización de PI3K y de las MAPK, que estimulan la proliferación, el crecimiento y la transformación celular, y que están implicadas en el desarrollo de cáncer (97).
La metformina reduce los niveles plasmáticos de glucosa al inhibir la gluconeogénesis en el hígado y aumentar la sensibilidad a la insulina y el consumo de glucosa en las células musculares. Se sugiere que esta regulación en el metabolismo radica en la capacidad de la metformina de inhibir del complejo I mitocondrial disminuyendo así la producción ATP y, en consecuencia, limitando los recursos energéticos necesarios para la proliferación de las células cancerosas. El desbalance AMP/ATP favorece la activación de la proteíncinasa activada por AMP (AMPK), la cual a su vez inhibe la síntesis de lípidos, ácidos grasos y la gluconeogénesis. Por lo tanto, a través de sus propiedades antidiabéticas (acción sistémica), la metfominna propicia un microambiente poco favorecedor para el crecimiento tumoral (98).
Varios estudios in vitro señalan que las biguanidas ejercen su acción antineoplásica directamente sobre las células de cáncer (acción celular), ya que son capaces de entrar a las células y modular varias vías de señalización que derivan en la inhibición de la proliferación y la inducción de muerte celular. Los estudios realizados por Wheaton et al., sugieren que los efectos antineoplásicas de la metformina requieren de la inhibición del complejo mitocondrial I (99). La inhibición de este complejo y la consecuente activación de AMPK, permite el bloqueo de la vía de mTORC1 (complejo 1 de la diana de rapamicina en células de mamífero), teniendo como resultado la inhibición de la síntesis de proteínas y del crecimiento celular. Además, la metformina también puede inhibir al complejo mTORC1 a través de la activación de RAG (GTPasas de Rag) y REDD1 (regulador 1 en el desarrollo y respuesta de daño del ADN), siendo esta una vía independiente de AMPK (98). La inhibición de mTORC1 disminuye a su vez la activación de los factores inducibles por hipoxia (HIF), lo cual deriva en la disminución de los mecanismos de supervivencia, metástasis y angiogénesis (98,100). Además, AMPK lleva a la activación de p53, disminuyendo la expresión de la ciclina D1 y consecuentemente induce el arresto y la muerte celular por apoptosis y autofagia (98). Asimismo, la AMPK reduce los procesos inflamatorios a través de la inhibición de NFκB y la disminución de la IL-6 e IL-8 (98,101).
Ensayos realizados en varios modelos animales muestran que la metformina reduce el crecimiento de los tumores de cáncer de mama (102), colorrectal (103), pulmón (104,105), páncreas (106,107) y ovario (108). Algunos estudios epidemiológicos sugieren una disminución en la incidencia de neoplasias y un aumento en la supervivencia de los pacientes diabéticos con cáncer que son tratados con metformina, a diferencia de pacientes tratados con sulfonilurea e insulina o incluso personas no diabéticas (109-111), encontrándose un posible efecto dosis-respuesta (112). El metaanálisis dirigido por De Censi et al., muestra que el tratamiento con metformina está asociado a una disminución del 30% en el riesgo de incidencia de cáncer en pacientes con diabetes tipo 2, además de observarse tendencias en la reducción de la mortalidad en todos los cánceres, en especial el cáncer de páncreas y hepatocelular (109). Estos análisis retrospectivos han dado pie a numerosos estudios clínicos que pudieran corroborar la posible eficacia de la metformina en el tratamiento del cáncer. Sin embargo, muchos de ellos han arrojado resultados inconclusos o contradictorios; dificultades como el fácil acceso de los pacientes a este medicamento y la posible disparidad entre las dosis administradas en pacientes diabéticos y la dosis requerida para lograr un efecto antineoplásico, pueden alterar las conclusiones de los estudios clínicos.
El papel de la metformina contra el cáncer no solo se centra en su potencial antineoplásico per se, sino que podría desempeñar un efecto adyuvante; sin embargo, algunos estudios muestran variaciones en los resultados. Pruebas in vitro han mostrado que la metformina mejora la efectividad de diferentes tratamientos, aumentando la sensibilidad a la radiación de las células de cáncer de mama MCF-7 al potenciar la activación de AMPK (113), y la apoptosis inducida por paclitaxel y cisplatino en células de cáncer endometrial (114). No obstante, el uso de esta biguanida en combinación con otros agentes quimioterapéuticos debe ser bien evaluado, ya que puede actuar como un antagonista del cisplatino, promoviendo la quimiorresistencia de líneas celulares de glioma, neuroblastoma, fibrosarcoma leucemia y carcinoma de células escamosas de esófago (101,115,116).
CONCLUSIONES
En la última década han aumentado los estudios sobre la obesidad y su relación en el desarrollo de cáncer. Desde el siglo pasado se sabe que el exceso de grasa corporal conlleva a un sinfín de enfermedades crónicas que acompañan a la obesidad, con lo cual se incrementa de manera considerable la morbilidad y la mortandad. Sin embargo, no se consideraba a la obesidad como un factor de riesgo en el desarrollo de cáncer, la evidencia actual nos permite entender que el exceso de tejido adiposo puede producir hormonas en cantidades suficientes que inducen cambios metabólicos y modulan varias vías de señalización que fomentan la proliferación y supervivencia celular, además de beneficiar la inflamación crónica subclínica y la formación de radicales libres que, en conjunto, favorecen el desarrollo de diversos tipos de cáncer.
Contribuciones de autoría: DHC y CFP participaron en la concepción, diseño del artículo e imágenes. GACA y MEH particparon en la revisión crítica del artículo. GEAA y JMD particparon en la recolección de datos. DHC y GACA particparon en la redacción del artículo y en la aprobación de la versión final.
Fuentes de financiamiento: este proyecto no contó con ninguna fuente de financiamiento.
Conflictos de interés: los autores declaran no tener conflicto de interés.
Referencias Bibliográficas
1. Kumar V, Aster J, Fausto N, Abbas A. Robbins y Cotran. Patología estructural y funcional. 8a ed. Philadelphia: Elsevier Saunders. [ Links ] 2011.
2. Organización Mundial de la Salud. Obesidad y sobrepeso. Nota descriptiva N°311 2015 [Internet]. Ginebra: OMS; 2015 [citado el 8 de enero de 2015]. Disponible en: http://www.who.int/mediacentre/factsheets/fs311/es/ [ Links ]
3. Food and Agriculture Organization, United Nations. Food and nutrition in numbers. Roma: FAO; 2014. [ Links ]
4. Cirilo B. Aspectos epidemiológicos de la obesidad en América Latina: los desafíos a futuro. Medwave. 2012;12(1): e5289 doi: 10.5867/medwave.2012.01.5289 [ Links ]
5. Organización Mundial de la Salud. Apartado. Temas de salud, Cáncer [Internet]. Ginebra: OMS; 2014 [citado el 8 de enero de 2015]. Disponible en: http://www.who.int/topics/ cancer/es/ [ Links ]
6. Organización Mundial de la Salud. Cáncer. Datos y cifras. Nota descriptiva N°297 [Internet]. Ginebra: OMS; 2015 [citado el 8 de enero de 2015]. Disponible en: http://www.who.int/mediacentre/factsheets/fs297/es/ [ Links ]
7. Tierney L, McPhee S, Papadakis M. Diagnóstico clínico y tratamiento. 35 ed. México, DF: Manual Moderno; 2000. p. 535-636. [ Links ]
8. Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981;66(6):1191-308. [ Links ]
9. Rao A, Agarwal S. Role of lycopene as antioxidante carotenoid in the prevention of chronic diseases: a review. Nutrition research. 1999;19(2):305-23. doi:10.1016/S0271- 5317(98)00193-6 [ Links ]
10. Snowdon DA, Phillips RL, Choi W. Diet, obesity, and risk of fatal prostate cancer. Am J Epidemiol. 1984;120(2):244-50. [ Links ]
11. Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86(3):556-65. [ Links ]
12. Hano O, Wood L, Villa O. Obesidad y riesgo de cáncer colorrectal. Rev Cubana Invest Biomed. 2011;30(2):251-9. [ Links ]
13. Forga L, Petrina E, Barbería J. Complicaciones de la obesidad. ANALES Sis San Navarra. 2002;25 Supl1:117-26. [ Links ]
14. Reforzo-Membrives J, Rocca DZ. Metabolism of sexual hormones in obesity. Rev Arg Endocrinol Metab. 1989;26(1):23-7. [ Links ]
15. Aguilar Cordero MJ, González Jiménez E, García López AP, Álvarez Ferré J, Padilla López CA, Guisado Barrilao R, et al. Obesidad y su implicación en el cáncer de mama. Nutr Hosp. 2011;26(4):899-903. doi: 10.1590/S0212-16112011000400033. [ Links ]
16. Haakinson DJ, Leeds SG, Dueck AC, Gray RJ, Wasif N, Stucky CC, et al. The impact of obesity on breast cancer: a retrospective review. Ann Surg Oncol. 2012;19(9):3012-8. doi: 10.1245/s10434-012-2320-8 [ Links ]
17. Jung S, Egleston BL, Chandler DW, Van Horn L, Hylton NM, Klifa CC, et al. Adolescent endogenous sex hormones and breast density in early adulthood. Breast Cancer Res. 2015;17:77. doi: 10.1186/s13058-015-0581-4. [ Links ]
18. McCormack VA, dos Santos Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: a metaanalysis. Cancer Epidemiol Biomarkers Prev. 2006;15(6):1159-69. [ Links ]
19. Cordero-Aguilar MJ, González Jiménez E, García López P, Alvarez Ferre J, Padilla López CA. Obesidad y niveles séricos de estrógenos; importancia en el desarrollo precoz del cáncer de mama. Nutr Hosp. 2012;27(4):1156-9. doi: 10.3305/nh.2012.27.4.5854. [ Links ]
20. Van den Broeck T, Tosco L, Prekovic S, Joniau S. Obesity and prostate cancer research. Cent European J Urol. 2014;66(4):428-9. doi: 10.5173/ceju.2013.04.art8. [ Links ]
21. Morales Clavijo M, Carvajal Garcés CF. Obesity and leptin resistance. Gac Med Bol. 2010;33(1):63-8. [ Links ]
22. Sinha YN, Salocks CB, Vanderlaan WP. Control of prolactin and growth hormone secretion in mice by obesity. Endocrinology. 1976;99(3):881-6. [ Links ]
23. Rosmond R, Dallman MF, Bjorntorp P. Stress-related cortisol secretion in men: relationships with abdominal obesity and endocrine, metabolic and hemodynamic abnormalities. J Clin Endocrinol Metab. 1998;83(6):1853-9. [ Links ]
24. Stolzenberg-Solomon RZ, Newton CC, Silverman DT, Pollak M, Nogueira LM, Weinstein SJ, et al. Circulating Leptin and Risk of Pancreatic Cancer: A Pooled Analysis From 3 Cohorts. Am J Epidemiol. 2015;182(3):187- 97. doi: 10.1093/aje/kwv041. [ Links ]
25. Herrera-Covarrubias D, Coria-Avila GA, Chavarria-Xicotencatl P, Fernandez-Pomares C, Manzo J, Aranda-Abreu GE, et al. Long-term administration of prolactin or testosterone induced similar precancerous prostate lesions in rats. Exp Oncol. 2015;37(1):13-8. [ Links ]
26. Gallagher EJ, LeRoith D. Obesity and Diabetes: The Increased Risk of Cancer and Cancer-Related Mortality. Physiol Rev. 2015;95(3):727-48. doi: 10.1152/ physrev.00030.2014. [ Links ]
27. Caprio S, Tamborlane WV, Silver D, Robinson C, Leibel R, McCarthy S, et al. Hyperleptinemia: an early sign of juvenile obesity. Relations to body fat depots and insulin concentrations. Am J Physiol. 1996;271(3 Pt 1):E626-30. [ Links ]
28. Almanza-Perez JC, Blancas-Flores G, Garcia-Macedo R, Alarcon-Aguilar FJ, Cruz M. [Leptin and its association with obesity and type 2 diabetes]. Gac Med Mex. 2008;144(6):535-42. [Article in Spanish] [ Links ]
29. Masuzaki H, Ogawa Y, Sagawa N, Hosoda K, Matsumoto T, Mise H, et al. Nonadipose tissue production of leptin: leptin as a novel placenta-derived hormone in humans. Nat Med. 1997;3(9):1029-33. [ Links ]
30. Hall J, Guyton A. Tratado de fisiología médica. 12a ed. Madrid: S.A. Elsevier España; 2011. [ Links ]
31. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395(6704):763-70. [ Links ]
32. Colli S, Silveira Cavalcante F, Peixoto Martins M, Sampaio FJ, da Fonte Ramos C. Leptin role in the rat prostate ventral lobe. Fertil Steril. 2011;95(4):1490-3. e1. doi: 10.1016/j. fertnstert.2010.12.029. [ Links ]
33. Tewari R, Rajender S, Natu SM, Goel A, Dalela D, Goel MM, et al. Significance of obesity markers and adipocytokines in high grade and high stage prostate cancer in North Indian men - a cross-sectional study. Cytokine. 2013;63(2):130-4. doi: 10.1016/j. cyto.2013.04.008. [ Links ]
34. Saglam K, Aydur E, Yilmaz M, Göktaş S. Leptin influences cellular differentiation and progression in prostate cancer. J Urol. 2003;169(4):1308-11. [ Links ]
35. Garofalo C, Surmacz E. Leptin and cancer. J Cell Physiol. 2006;207(1):12-22. [ Links ]
36. Tessitore L, Vizio B, Pesola D, Cecchini F, Mussa A, Argiles JM, et al. Adipocyte expression and circulating levels of leptin increase in both gynaecological and breast cancer patients. Int J Oncol. 2004 Jun;24(6):1529-35. [ Links ]
37. Weichhaus M, Broom J, Wahle K, Bermano G. Leptin inhibits proliferation of breast cancer cells at supraphysiological concentrations by inhibiting mitogenactivated protein kinase signaling. Oncol Lett. 2014;8(1):374-8. [ Links ]
38. Wei T, Lu L, Shen Q, Fang C. [Advances of the relationship between leptin system and non-small cell lung cancer]. Zhongguo Fei Ai Za Zhi. 2014;17(4):347-50. doi: 10.3779/j. issn.1009-3419.2014.04.10. [Article in Chinese] [ Links ]
39. Yabushita H, Iwasaki K, Obayashi Y, Wakatsuki A. Clinicopathological roles of adiponectin and leptin receptors in endometrial carcinoma. Oncol Lett. 2014;7(4):1109-17. [ Links ]
40. Zhou W, Tian Y, Gong H, Guo S, Luo C. Oncogenic role and therapeutic target of leptin signaling in colorectal cancer. Expert Opin Ther Targets. 2014;18(8):961-71. doi: 10.1517/14728222.2014.926889. [ Links ]
41. Ren H, Jia L, Zhao T, Zhang H, Chen J, Yang S, et al. Hypoxia inducible factor (HIF)- 1alpha directly activates leptin receptor (ObR) in pancreatic cancer cells. Cancer Lett. 2014. doi: 10.1016/j.canlet.2014.08.001. [ Links ]
42. Stattin P, Soderberg S, Hallmans G, Bylund A, Kaaks R, Stenman UH, et al. Leptin is associated with increased prostate cancer risk: a nested case-referent study. J Clin Endocrinol Metab. 2001;86(3):1341-5. [ Links ]
43. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117(Pt 8):1281-3. [ Links ]
44. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798- 809. doi: 10.1038/nrc2734. [ Links ]
45. Greenman Y, Tordjman K, Stern N. Increased body weight associated with prolactin secreting pituitary adenomas: weight loss with normalization of prolactin levels. Clin Endocrinol (Oxf ). 1998 May;48(5):547-53. [ Links ]
46. Carré N, Solomon G, Gertler A, Binart N. Effects of high affinity leptin antagonist on prolactin receptor deficient male mouse. PLoS One. 2014;9(3):e91422. doi: 10.1371/ journal.pone.0091422. [ Links ]
47. Mujagić Z, Srabović N, Mujagić H. The role of prolactin in human breast cancer. Biochemia Medica. 2009;19(3):236-49. doi. org/10.11613/BM.2009.022. [ Links ]
48. Freeman ME, Kanyicska B, Lerant A, Nagy G. Prolactin: Structure, Function, and Regulation of Secretion. Physiol Rev. 2000;80(4):1523-631. [ Links ]
49. Harper ME, Peeling WB, Cowley T, Brownsey BG, Phillips ME, Groom G, et al. Plasma steroid and protein hormone concentrations in patients with prostatic carcinoma, before and during oestrogen therapy. Acta Endocrinol (Copenh). 1976 Feb;81(2):409-26. [ Links ]
50. Saroff J, Kirdani RY, Chu TM, Wajsman Z, Murphy GP. Measurements of prolactin and androgens in patients with prostatic diseases. Oncology. 1980;37(1):46-52. [ Links ]
51. Nevalainen MT, Valve EM, Ingleton PM, Nurmi M, Martikainen PM, Harkonen PL. Prolactin and prolactin receptors are expressed and functioning in the human prostate. J Clin Invest. 1997;99(4):618-27. [ Links ]
52. McGown C, Birerdinc A, Younossi ZM. Adipose tissue as an endocrine organ. Clin Liver Dis. 2014;18(1):41-58. doi: 10.1016/j. cld.2013.09.012. [ Links ]
53. Carré N, Binart N. Prolactin and adipose tissue. Biochimie. 2014;97:16-21. doi: 10.1016/j.biochi.2013.09.023. [ Links ]
54. Brandebourg T, Hugo E, Ben-Jonathan N. Adipocyte prolactin: regulation of release and putative functions. Diabetes Obes Metab. 2007;9(4):464-76. [ Links ]
55. Kopelman PG. Physiopathology of prolactin secretion in obesity. Int J Obes Relat Metab Disord. 2000;24 Suppl 2:S104-8. [ Links ]
56. Leav I, Merk FB, Lee KF, Loda M, Mandoki M, McNeal JE, et al. Prolactin receptor expression in the developing human prostate and in hyperplastic, dysplastic, and neoplastic lesions. Am J Pathol. 1999;154(3):863-70. [ Links ]
57. Suttie A, Nyska A, Haseman JK, Moser GJ, Hackett TR, Goldsworthy TL. A grading scheme for the assessment of proliferative lesions of the mouse prostate in the TRAMP model. Toxicol Pathol. 2003;31(1):31-8. [ Links ]
58. Tuzcu A, Bahceci M, Dursun M, Turgut C, Bahceci S. Insulin sensitivity and hyperprolactinemia. J Endocrinol Invest. 2003;26(4):341-6. [ Links ]
59. Chirico V, Cannavó S, Lacquaniti A, Salpietro V, Mandolfino M, Romeo PD, et al. Prolactin in obese children: a bridge between inflammation and metabolicendocrine dysfunction. Clin Endocrinol (Oxf ). 2013 Oct;79(4):537-44. doi: 10.1111/cen.12183. [ Links ]
60. Ampuero J, Romero-Gomez M. Prevention of hepatocellular carcinoma by correction of metabolic abnormalities: Role of statins and metformin. World J Hepatol. 2015;7(8):1105-11. doi: 10.4254/ wjh.v7.i8.1105. [ Links ]
61. Olivares Reyes J, Arellano Plancarte A. Bases moleculares de las acciones de la insulina. REB 2008;27(1):9-18. [ Links ]
62. Björntorp P. Body fat distribution, insulin resistance, and metabolic diseases. Nutrition. 1997;13(9):795-803. [ Links ]
63. Björntorp P. Neuroendocrine factors in obesity. J Endocrinol. 1997;155(2):193-5. [ Links ]
64. Martínez SJ, Torres DPV, Juárez OMA. Los ácidos grasos y la lipotoxicidad: implicaciones metabólicas. Rev Fac de Med UNAM. 2013;56(1):5-18. [ Links ]
65. Giovannucci E. Insulin, insulin-like growth factors and colon cancer: a review of the evidence. J Nutr. 2001;131(11 Suppl):3109S- 20S. [ Links ]
66. Herrera-Covarrubias D, Fernández Pomares C, Aranda Abreu G, Dominguez Ortíz M, Hernández Aguilar M. The role of lycopene in prevention of prostate cancer. Eneurobiología. 2013;4(8):1-16. [ Links ]
67. Yu H, Spitz MR, Mistry J, Gu J, Hong WK, Wu X. Plasma levels of insulin-like growth factor-I and lung cancer risk: a case-control analysis. J Natl Cancer Inst. 1999;91(2):151- 6. [ Links ]
68. Mantzoros CS, Bolhke K, Moschos S, Cramer DW. Leptin in relation to carcinoma in situ of the breast: a study of premenopausal cases and controls. Int J Cancer. 1999;80(4):523-6. [ Links ]
69. Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998;351(9113):1393-6. [ Links ]
70. Liu X, Allen JD, Arnold JT, Blackman MR. Lycopene inhibits IGF-I signal transduction and growth in normal prostate epithelial cells by decreasing DHT-modulated IGF-I production in co-cultured reactive stromal cells. Carcinogenesis. 2008;29(4):816-23. [ Links ]
71. Baquedano E, Ruiz-Lopez AM, Sustarsic EG, Herpy J, List EO, Chowen JA, et al. The absence of GH signaling affects the susceptibility to high fat diet induced hypothalamic inflammation in male mice. Endocrinology. 2014;155(12):4856-67. doi: 10.1210/en.2014-1367. [ Links ]
72. Chae HW, Kim DH, Kim HS. Growth hormone treatment and risk of malignancy. Korean J Pediatr. 2015;58(2):41-6. doi: 10.3345/kjp.2015.58.2.41. [ Links ]
73. Bowers LW, Rossi EL, O’Flanagan CH, deGraffenried LA, Hursting SD. The Role of the Insulin/IGF System in Cancer: Lessons Learned from Clinical Trials and the Energy Balance-Cancer Link. Front Endocrinol (Lausanne). 2015;6:77. doi: 10.3389/ fendo.2015.00077. [ Links ]
74. Geneser F. Histología sobre bases biomoleculares. 3rd ed. Madrid, España: Editorial Medica Panamericana; 2000. p. 813. [ Links ]
75. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796-808. [ Links ]
76. Zhang L, Fan XM. The pathological role of microRNAs and inflammation in colon carcinogenesis. Clin Res Hepatol Gastroenterol. 2015;39(2):174-9. doi: 10.1016/j.clinre.2014.06.013. [ Links ]
77. Porcaro AB, Novella G, Molinari A, Terrin A, Minja A, De Marco V, et al. Prostate volume index and chronic inflammation of the prostate type IV with respect to the risk of prostate cancer. Urol Int. 2015;94(3):270-85. doi: 10.1159/000362176. [ Links ]
78. Masson-Lecomte A, Rava M, Real FX, Hartmann A, Allory Y, Malats N. Inflammatory biomarkers and bladder cancer prognosis: a systematic review. Eur Urol. 2014;66(6):1078-91. doi: 10.1016/j. eururo.2014.07.033. [ Links ]
79. Lanaya H, Natarajan A, Komposch K, Li L, Amberg N, Chen L, et al. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat Cell Biol. 2014;16(10):972-81, 1-7. doi: 10.1038/ncb3031. [ Links ]
80. Perez-Hernandez AI, Catalán V, GómezAmbrosi J, Rodríguez A, Frühbeck G. Mechanisms linking excess adiposity and carcinogenesis promotion. Front Endocrinol (Lausanne). 2014;5:65. doi: 10.3389/ fendo.2014.00065. [ Links ]
81. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821-30. [ Links ]
82. Coll-García E. Lipotoxicidad y resistencia a la insulina. Mecanismos moleculares de la insulinoresistencia. Infor Med. 2002;4(6):399-418. [ Links ]
83. Feingold KR, Grunfeld C. Role of cytokines in inducing hyperlipidemia. Diabetes. 1992;41 Suppl 2:97-101. [ Links ]
84. Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab. 1998;83(3):847-50. [ Links ]
85. Allavena P, Garlanda C, Borrello MG, Sica A, Mantovani A. Pathways connecting inflammation and cancer. Curr Opin Genet Dev. 2008;18(1):3-10. doi: 10.1016/j. gde.2008.01.003. [ Links ]
86. Shen HM, Tergaonkar V. NFkappaB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis. 2009;14(4):348-63. doi: 10.1007/s10495-009-0315-0. [ Links ]
87. Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAILmediated tumor regression. Cancer Cell. 2004;6(3):297-305. [ Links ]
88. Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8(1):33-40. doi: 10.1038/nrd2781. [ Links ]
89. Yu JH, Kim H. Oxidative stress and cytokines in the pathogenesis of pancreatic cancer. J Cancer Prev. 2014;19(2):97-102. doi: 10.15430/JCP.2014.19.2.97. [ Links ]
90. Betteridge DJ. What is oxidative stress? Metabolism. 2000;49(2 Suppl 1):3-8. [ Links ]
91. Laguna J, Piña-Garza E. Bioquímica de Laguna. 6a ed. México, DF: Manual Moderno; 2007. [ Links ]
92. van Dam PS, Bravenboer B, van Asbeck BS, van Oirschot JF, Marx JJ, Gispen WH. Effects of insulin treatment on endoneurial and systemic oxidative stress in relation to nerve conduction in streptozotocin-diabetic rats. Eur J Clin Invest. 1996;26(12):1143-9. [ Links ]
93. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114(12):1752-61. [ Links ]
94. Chandel N. Four key questions about metformin and cancer. BMC Biol. 2014;12:85. doi: 10.1186/s12915-014- 0085-1. [ Links ]
95. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309-14. [ Links ]
96. DeCensi A, Gennari A. Insulin breast cancer connection: confirmatory data set the stage for better care. J Clin Oncol. 2011 Jan 1;29(1):7- 10. doi: 10.1200/JCO.2010.32.3022. [ Links ]
97. Quinn BJ, Kitagawa H, Memmott RM, Gills JJ, Dennis PA. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab. 2013 Sep;24(9):469-80. doi: 10.1016/j.tem.2013.05.004. [ Links ]
98. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242. doi: 10.7554/ eLife.02242. [ Links ]
99.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721- 32. [ Links ]
100.Pernicova I, Korbonits M. Metformin-- mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10(3):143-56. doi: 10.1038/nrendo.2013.256. [ Links ]
101.Gao S, Jiang J, Li P, Song H, Wang W, Li C, et al. Attenuating tumour angiogenesis: a preventive role of metformin against breast cancer. Biomed Res Int. 2015;2015:592523. doi: 10.1155/2015/592523. [ Links ]
102.Tomimoto A, Endo H, Sugiyama M, Fujisawa T, Hosono K, Takahashi H, et al. Metformin suppresses intestinal polyp growth in ApcMin/+ mice. Cancer Sci. 2008;99(11):2136-41. doi: 10.1111/j.1349-7006.2008.00933.x. [ Links ]
103.Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen- -induced lung tumorigenesis. Cancer Prev Res (Phila). 2010;3(9):1066-76. doi: 10.1158/1940-6207.CAPR-10-0055. [ Links ]
104.Antonoff MB, D’Cunha J. Teaching an old drug new tricks: metformin as a targeted therapy for lung cancer. Semin Thorac Cardiovasc Surg. 2010;22(3):195-6. doi: 10.1053/j.semtcvs.2010.10.015. [ Links ]
105.Mohammed A, Janakiram NB, Brewer M, Ritchie RL, Marya A, Lightfoot S, et al. Antidiabetic Drug Metformin Prevents Progression of Pancreatic Cancer by Targeting in Part Cancer Stem Cells and mTOR Signaling. Transl Oncol. 2013;6(6):649-59. [ Links ]
106.Aljada A, Mousa SA. Metformin and neoplasia: implications and indications. Pharmacol Ther. 2012 Jan;133(1):108-15. doi: 10.1016/j.pharmthera.2011.09.004. [ Links ]
107.Rattan R, Graham RP, Maguire JL, Giri S, Shridhar V. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia. 2011;13(5):483-91. [ Links ]
108.Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila). 2010;3(11):1451-61. doi: 10.1158/1940-6207.CAPR-10-0157. [ Links ]
109.Currie CJ, Poole CD, Jenkins-Jones S, Gale EA, Johnson JA, Morgan CL. Mortality after incident cancer in people with and without type 2 diabetes: impact of metformin on survival. Diabetes care. 2012;35(2):299-304. doi: 10.2337/dc11-1313. [ Links ]
110.Kasznicki J, Sliwinska A, Drzewoski J. Metformin in cancer prevention and therapy. Ann Transl Med. 2014;2(6):57. doi: 10.3978/j.issn.2305-5839.2014.06.01. [ Links ]
111.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. Bmj. 2005;330(7503):1304-5. [ Links ]
112.Song CW, Lee H, Dings RP, Williams B, Powers J, Santos TD, et al. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci Rep. 2012;2:362. doi: 10.1038/srep00362. [ Links ]
113.Dong L, Zhou Q, Zhang Z, Zhu Y, Duan T, Feng Y. Metformin sensitizes endometrial cancer cells to chemotherapy by repressing glyoxalase I expression. J Obstet Gynaecol Res. 2012;38(8):1077-85. doi: 10.1111/j.1447-0756.2011.01839.x. [ Links ]
114.Janjetovic K, Vucicevic L, Misirkic M, Vilimanovich U, Tovilovic G, Zogovic N, et al. Metformin reduces cisplatin-mediated apoptotic death of cancer cells through AMPK-independent activation of Akt. Eur J Pharmacol. 2011;651(1-3):41-50. doi: 10.1016/j.ejphar.2010.11.005. [ Links ]
115.Damelin LH, Jivan R, Veale RB, Rousseau AL, Mavri-Damelin D. Metformin induces an intracellular reductive state that protects oesophageal squamous cell carcinoma cells against cisplatin but not copper-bis(thiosemicarbazones). BMC cancer. 2014;14:314. doi: 10.1186/1471- 2407-14-314. [ Links ]
Correspondencia: Deissy Herrera-Covarrubias
Dirección: Centro de Investigaciones Cerebrales (CICE). Av. Luis Castelazo Ayala s/n, Colonia Industrial Las Ánimas. Xalapa, Ver. México, C.P. 91193.
Teléfono: 01 (228) 841 89 00 Ext. 13616.
Correo electrónico: dherrera@uv.mx
Recibido: 03-04-15
Aprobado: 08-07-15
