










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
Print version ISSN 1726-4634
Rev. perú. med. exp. salud publica vol.33 no.2 Lima Apr./Jun. 2016
http://dx.doi.org/10.17843/rpmesp.2016.332.2192
ARTÍCULO ORIGINAL
Análisis genómico comparativo de cepas peruanas de Mycobacterium tuberculosis
Comparative genomic analysis of peruvian strains of Mycobacterium tuberculosis
David Tarazona 1,a,c , Marco Galarza1,a, Kelly S. Levano1,b, Heinner Guio1,d
1 Laboratorio de Referencia Nacional de Biología Molecular y Biotecnología, Centro Nacional de Salud Pública, Instituto Nacional de Salud, Lima, Perú.
a Biólogo molecular; b PhD en Bioquímica; c genetista; d PhD en Ciencias Médicas
RESUMEN
Objetivos. Analizar comparativamente tres secuencias genómicas de Mycobacterium tuberculosis(MTB): INS-SEN,cepa sensible; INS-MDR, cepa multidrogorresistente e INS-XDR, cepa extensamente resistente, procedentes de la Ciudad de Lima, Perú. Materiales y métodos. Se identificaron los polimorfismos de un solo nucleótido (SNPs) específicos en las cepas INS-SEN, INS-MDR y INS-XDR mediante el criterio de inclusión/exclusión. Se compararon los tres genomas de MTB y se construyó una filogenia molecular con 27 cepas de MTB de otros estudios, disponibles de la base de datos Genbank. Los SNPs específicos en cada genoma fueron organizados en clústers de grupos ortólogos (COGs). Resultados. El análisis de genomas permitió identificar un conjunto de SNPs asociados a determinantes de virulencia (familia de proteínas mce, policetidos, phiRv1, transposasas, metiltransferasas y relacionados a síntesis de vitaminas) principalmente. Se observa una estrecha relación entre la cepa INS-MDR y INS-XDR, con solo un 6,1% de SNPs diferentes, sin embargo, la cepa INS-SEN presenta un 50,2 y 50,3% de SNPs diferentes a las cepas MDR y XDR, respectivamente. La filogenia molecular agrupó a las cepas peruanas dentro del linaje LAM y cercanamente a las cepas F11 y KZN de Sudáfrica. Conclusiones. Se evidenció una alta similitud (99,9%) de la cepa INS-SEN con la cepa sudafricana F11, de gran alcance mundial, mientras los análisis de las cepas INS-MDR e INS-XDR demuestran una probable expansión de la familia KZN, cepa de Sudáfrica con alta virulencia y patogenicidad.
Palabras clave: Tuberculosis; Genómica; Farmacorresistencia bacteriana (fuente: DeCS BIREME).
ABSTRACT
Objectives. To comparatively analyze three genomic sequences of Mycobacterium tuberculosis(MTB), including sensitive (INS-SEN), multi-drug-resistant (INS-MDR), and extremely drug-resistant (INS-XDR) strains, collected in Lima, Peru. Materials and Methods. Specific single nucleotide polymorphisms (SNPs) were identified in the INS SEN, INS-MDR, and INS-XDR strains according to the inclusion/exclusion criteria. The three MTB genomes were compared and a molecular phylogeny was constructed with 27 MTB strains from other studies available from the Genbank database. Results. The specific SNPs in each genome were organized in clusters of orthologous groups (COGs). The genomic analysis allowed for the identification of a set of SNPs associated mainly with virulence determinants (family of mce proteins, polyketides, phiRv1, transposase, and methyltransferases, and other related to vitamin synthesis). A close correlation between the INS-MDR and INS-XDR strains was observed, with only a 6.1% difference in SNPs; however, the INS-SEN strain had 50.2% and 50.3% different SNPs from the MDR and XDR strains, respectively. The molecular phylogeny grouped the Peruvian strains within the LAM lineage and closely to the F11 and KZN strains from South Africa. Conclusions. High similarity (99.9%) was noted between the INS-SEN strain and the F11 South African strain with broadglobal scope, while the analysis of the INS-MDR and INS-XDR strains showed a likely expansion of the KZN family, a South African strain with high virulence and pathogenicity.
Key words: Tuberculosis; Genomic; Drug Resistant, Bacterial (source: MeSH NLM).
INTRODUCCIÓN
Según la Organización Mundial de la Salud (OMS), durante el 2013 se han reportado 6,1 millones de casos de tuberculosis (TB). De estos, el 3,5% correspondieron a casos nuevos de tuberculosis multidrogorresistente (TB-MDR) y el 9% de estos casos han constituido casos nuevos de tuberculosis extensamente resistente (TB-XDR). En Perú, la alta carga de TB y el incremento de cepas multidrogorresistentes (MDR) es uno de los mayores problemas en salud pública. En el año 2013, la tasa de incidencia de TB en Perú fue de 124/100 000 habitantes, de los cuales un 3,9% (850) fueron casos nuevos de TB-MDR (1).
Si bien los casos nuevos de TB han disminuido en los últimos años, los casos de TB-MDR se han incrementado en los últimos 3 años. Durante el año 2014, Perú representó el 41,3% de todos los casos TB-MDR de la región de las Américas (2). Lima concentra el 80 y 92% de casos TB-MDR y TB-XDR respectivamente (3), donde un factor importante es el transporte público peruano, como contribuyente de factor de riesgo para TB y su diseminación (4).
La TB-MDR está definida como la resistencia a rifampicina (RIF) e isoniacida (INH), dos de las drogas más potentes usadas en el tratamiento antituberculoso.
La TB-XDR definida como la resistencia a RIF, INH, fluoroquinolonas y, al menos, uno de los antibióticos inyectables (amikacina, kanamicina o capreomicina).
La RIF es un antibiótico bactericida que inhibe la enzima ARN polimerasa de la micobacteria. La región genética relacionada a resistencia a RIF está localizada en una región de 81 pb del gen rpoB de Mycobacterium tuberculosis (MTB). La INH es una prodroga que esactivada por la enzima katG (catalasa peroxidasa) de MTB, y actúa inhibiendo la síntesis de ácido micólico. La resistencia a INH es frecuentemente observada en el codón 315 del gen katG, además de otras importantes regiones de mutación que pueden ocurrir en los genes ahpC, kasA, ndh y la región promotora inhA (5).
Existe una alta diversidad genética de cepas de MTB a nivel mundial que causan la enfermedad. Se han descrito como prevalentes en el mundo los siguientes genotipos: Haarlem (H), Latin América-Mediterráneo (LAM), T, Este de África-India (EAI), Central-Asia (CAS), X y Beijing. Cada nombre está atribuido al lugar de procedencia donde se identificó inicialmente. Asimismo, los recientes estudios en análisis de genomas están identificando nuevas regiones en MTB como posibles sitios polimórficos en relación a resistencia a drogas antituberculosas (6).
La evolución de cepas multidrogorresistentes (MDR) y extensamente resistente (XDR) tiene una estrecha relación a cepas provenientes de Sudáfrica, como las familias F11 y KZN. La familia F11 data de los años 1992- 1998, provenientes del oeste de Cape en Sudáfrica y que representó el 21,4% de todos los casos de TB en esos años. Presentan como características principales: la presencia de bandas patrones de la región IS6110 que varía de 11 a 19 bandas, ausencia de regiones espaciadoras 9 a 11; 21 a 24 y 33 a 36, así como la presencia de polimorfismo C491T en el gen rrs (7) . Por otro lado, la familia KZN proviene de la región de KwaZulu-Natal en Sudáfrica, y data de un brote de los años 2005-2007. Es una cepa altamente virulenta que causó 52 muertes de 53 pacientes con diagnóstico TB-XDR y HIV (8)
Existe evidencia que poblaciones nativas peruanas han sido afectados por TB (9). Sin embargo, el genotipo de MTB que los afecta aún permanece desconocido. Se tiene registrado que en Perú existen familias genéticas Beijing desde la llegada de migrantes asiáticos en el decenio de 1900 (10), y que recientemente fue reportado genotipos similares en España e Italia (11). El primer estudio de la diversidad genética de MTB en el Perú de aislados sensible y drogorresistentes, indica que los genotipos predominantes son: LAM (23,8%), T (23,8%), Haarlem (22,3%) y Beijing (9,3%) (12). Sheen et al. reportan como prevalentes a los linajes Haarlem(28,7%), LAM (28,3%) y T (20,3%) en pacientes con TB y VIH (13). Investigadores del Instituto Nacional de Salud (INS) de Perú, identificaron que pacientes con TB-XDR presentaron los linajes Haarlem (43,6%), T (27,7%), LAM (16,2%) y Beijing (9,5%) (3). Estos estudios permiten una primera aproximación, de los genotipos LAM, Haarlem y T en Perú. Grandjean et al. (2015), en un estudio con 2139 aislados, encontraron que la familia LAM puede representar al menos el 50% de todos los casos de resistencia a droga en la región peruana (14).
Durante el 2014, se realizó el secuenciamiento completo de tres genomas de MTB (sensible, MDR y XDR) aislados en Perú, de linaje LAM. El presente estudio tiene como objetivo realizar un análisis comparativo de los tres genomas frente a otros genomas referenciales, y poder establecer su relación genética entre ellos.
MATERIALES Y MÉTODOS
CEPAS, SENSIBILIDAD A DROGAS Y PURIFICACIÓN DE ADN
Los aislados fueron cultivados en medio solido Löwenstein-Jensen y la susceptibilidad a drogas de primera y segunda línea fue por el método estándar Agar Proporciones en Placa (APP), en el Laboratorio de Referencia Nacional deMicobacterias del Instituto Nacional de Salud. Se realizó la extracción de ADN usando el kit PureLink® Genomic DNA (Invitrogen), y la cuantificación del ADN se hizo en el equipo NanoDrop 8000 (Thermo Scientific) del Laboratorio de Referencia Nacional de Biotecnología y Biología Molecular del INS. En la Tabla 1 se muestran las características básicas de los aislados de MTB sensible, multidrogorresistente y extremadamente resistente, seleccionados para el estudio. Mediante la metodología MIRU-VNTR (Mycobacterial Interspersed Repetitive Unit-Variable Number Of Tandem Repeat) se identificó el linaje de MTB.
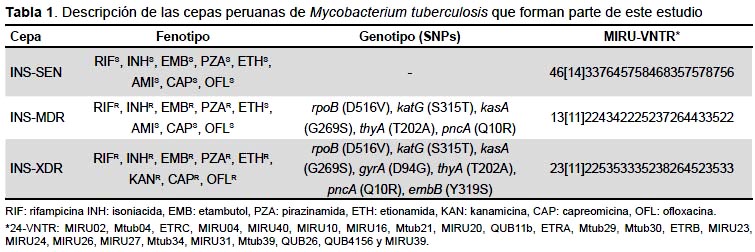
DATOS GENÓMICOS DE CEPAS DE Mycobacterium tuberculosis
Se construyeron librerías genómicas de extremos pareados, las cuales fueron secuenciadas en el equipo HiSeq 2000, Illumina (Macrogen, Korea)(15-17), siguiendo los protocolos del fabricante. El ensamblaje de la secuenciación de lecturas se llevó a cabo por el software BWA versión v 0.5.9-r16, usando el genoma referencial H37Rv.
ANÁLISIS DE SECUENCIAS Y FILOGENIA MOLECULAR
Determinamos los SNPs estrechamente relacionados a MTB H37Rv (AL123456.2) mediante los programas SamTools, SNPtree y scripts en Perl para INS-SEN (15), INS-MDR (16) e INS-XDR (17). La anotación de secuencias codificantes en INS-SEN, INS-MDR y INS-XDR fue realizada usando los programas RAST (Rapid Annotations using Subsystem Technology) y PGAAP (Prokaryotic Genome Annotation Pipeline). Adicionalmente, las proteínas se agruparon en clúster de grupos ortólogos (COG) mediante BLAST y se organizaron los SNP en COG.
Se realizó un análisis comparativo por criterio de inclusión/exclusión usando un total de 785, 805 y 814 SNPs de INS-SEN, INS-MDR y INS-XDR, respectivamente, empleando las bases de datos, y elaboradas con pipelines en Perl - Linux. Adicionalmente, se elaboró una filogenia por Máximum Likelihood de los genomas de MTB listados en la Tabla 2, para ello se utilizó el programa FastTree y se utilizó el programa de graficas FigTree.
RESULTADOS
ESTRUCTURA DE GENOMAS DE MTB EN AISLADOS PERUANOS
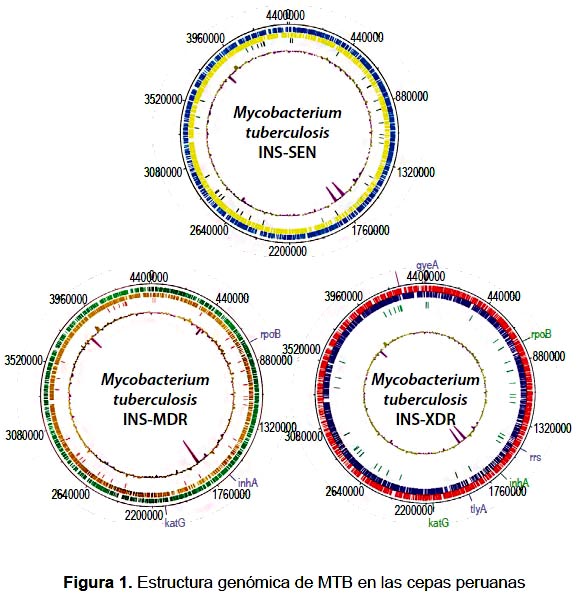
INS-SEN (NCBI: JAQH01000000) tuvo 61 422 158 lecturas de secuencias con 1406x de cobertura genómica logrando un tamaño de 4 383 671 pb y un contenido GC de 65,6%. INS-MDR (NCBI: JAQI010000000) de 58 157 302 lecturas de secuencias con 1331x de cobertura logrando un tamaño de 4383 671 pb y un contenido GC de 65,6%. INS-XDR (NCBI: JANH01000000) de 49 793 402 lecturas de secuencias con 1140x de cobertura logrando un tamaño de 4 391 020 pb y un contenido GC de 65,4%.
En nuestro análisis, encontramos que los tres genomas INS-SEN, INS-MDR y INS-XDR tienen 4389 secuencias codificantes, 45 tRNA, 03 tRNA (Figura 1). El estudio comparativo de cada genoma frente a H37Rv encontró que INS-SEN, INS-MDR y INS-XDR tienen 499, 805 y 815 SNPs, respectivamente. El porcentaje de SNPs localizados en regiones codificantes fueron, para INS-SEN (88,2%), INS-MDR (87,3%) y INS-XDR (87,3%) aproximadamente.
COMPARACIÓN DE SECUENCIAS DE MTB EN AISLADOS PERUANOS
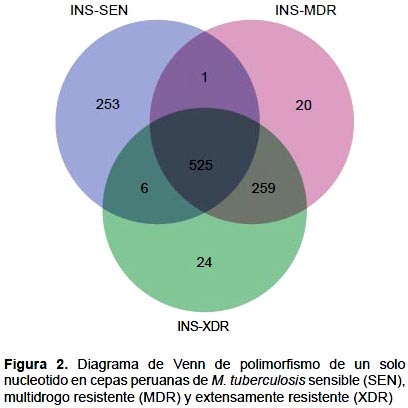
La comparación de genomas de MTB de familia LAM por el criterio de inclusión/exclusión determinó que las cepas INS-SEN, INS-MDR y INS-XDR tienen SNPs específicos y compartidos (Figura 2). El número de SNPs propios fueron 253 para INS-SEN, 20 para INS-MDR y 24 para INS-XDR. Los SNPs compartidos para los tres genomas es de 48,3% (525/1088). El número de SNPs compartidos entre INS-SEN y INS-MDR es de 26,2%; INS-SEN y INS-XDR 26,4%.
FILOGENIA MOLECULAR DE CEPAS DE MTB
La filogenia molecular construida a partir de 30 genomas de MTB de familias representativas de linajes LAM, Beijing, CAS, Haarlem, EAI y Ural (Figura 3) ha mostrado ser eficaz para bacterias genéticamente monomórficos como MTB. La filogenia agrupó los aislados en linajes, sin embargo no se puede observar grupos bien diferenciados de cepas sensibles o drogo resistentes. Esta filogenia divide todos los aislados en diferentes grupos, estableciendo que INS-SEN se encuentra en el linaje LAM y más relacionado a HM y F11 (proveniente del brote en Cape en Sudáfrica).
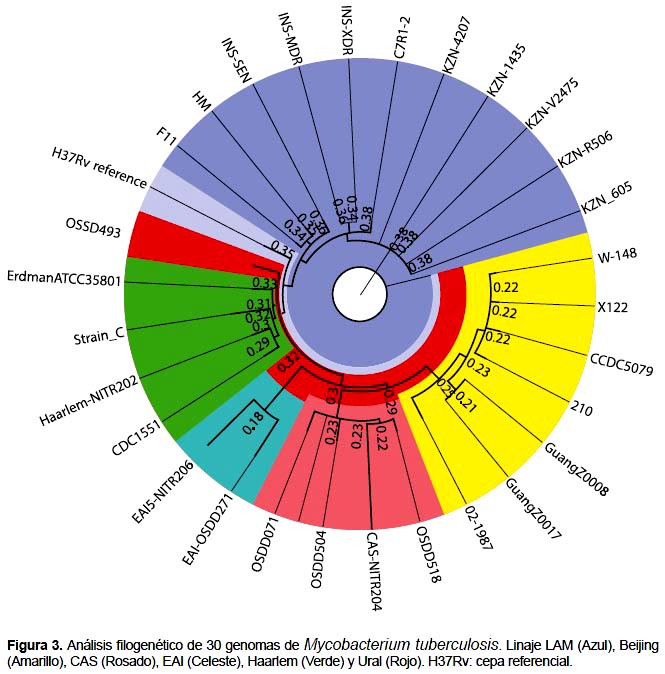
En contraste los aislados, INS-MDR e INS-XDR también se encuentran en linaje LAM pero más relacionados a la familia KZN (proveniente del brote KwaZulu-Natal en Sudáfrica).
ANÁLISIS DE GRUPOS ORTÓLOGOS (COGS)
Los genomas en este estudio fueron: sensible, MDR, XDR y de linaje LAM. Se formó una lista de SNPs individual para cada genoma INS frente a H37Rv. Las proteínas codificadas que presentaron SNPs fueron asociadas a veinte clases funcionales basados en grupos de ortólogos (COG). Los SNPs se normalizaron con respecto al número de genes de un determinado COG en MTB y con el número total de SNPs identificados (Figura 4). Los grupos K: transcripción; J: traducción, biogénesis ribosomal; O: modificación postraduccional, chaperonas; V: mecanismo de defensa; U: secreción, tráfico intracelular y transporte vesicular, y A: procesamiento y modificación de ARN; no tuvieron un ajuste estructural sobre la asignación, lo que sugiere que los genes de estas categorías podrían estar bajo presión evolutiva. En todas las cepas peruanas, los números significativos de SNPs se encuentran en las proteínas Q: biosíntesis, transporte y catabolismo de metabolismo secundario y L: replicación, recombinación y reparación. Dentro del grupo Q se encuentran los genes de mce y síntesis de policetidos. Mientras que en el grupo L encontramos genes de la ADN girasa, fago phiRv1, transposasas y ligasas, también metiltransferasas, más exclusivamente en las micobacterias drogorresistentes. El grupo H: metabolismos y transporte de coenzima, en la cepa sensible ha mostrado mayor valor significativo que las drogorresistentes (MDR y XDR), donde encontramos los genes que codifican enzimas para síntesis de riboflavina, cobalamina, glutamina, molibdeno, nicotidamina y magnesio quelatasa. Notamos también que el grupo G – metabolismo y transporte de carbohidratos es significativamente diferente entre la cepa sensibles y las drogorresistentes, dentro de este grupo de proteínas encontramos a las transcetolasas, opcA, transportador de azúcar, glucosidasas, permeasas de la familia MFS.
DISCUSIÓN
En la actualidad, los avances en genómica comparativa permiten conocer en forma precisa, los polimorfismos relacionados a la fisiología, factores de virulencia y resistencia a drogas de los principales agentes infecciosos de enfermedades con alto impacto en salud pública. En este estudio se reporta por primera vez un análisis de tres genomas de MTB (sensible, MDR y XDR) que provienen de pacientes que solo han vivido en la ciudad de Lima, Perú, por lo que es razonable pensar que estos organismos representan una fracción de las cepas sensibles y drogorresistentes que circulan en Perú. En general, los aislados del MTB (INS-SEN, INS-MDR e INS-XDR), muestran un conjunto de SNP más asociados a los grupos funcionales Q y L, la familia Q está más representado por la familia de proteína Mce, un factor de virulencia importante en MTB. Tienen funciones en la inducción tráfico de substrato en la fase de infección (18), modificación del perfil lipídico y capacidad de supervivencia intracelular (19). La disrupción funcional de Mce1 causa hipervirulencia mientras las disrupciones en Mce2 y Mce3 confieren que los aislados sean atenuados (20). En este estudio seevidenció que INS-SEN contiene una mutación adicional en Ser270Asn de Mce2D lo que podría relacionarse a su virulencia y atenuación. Adicionalmente, se encontró Val87Ala de Mce1B correspondiente solo a los genomas INS-MDR y INS-XDR que podrían ser los responsables de los cambios lipídicos en la membrana.
Mientras que en la familia L encontramos a los fagos PhiRv1, que tienen un sistema activo de integración/ escisión que les da facultades de cambiar de posiciones en el genoma de MTB (21). El aislado INS-SEN ha mostrado más variabilidad en secuencias PhiRv1 que en los aislados drogorresistentes. Aunque no se ha reportado asociaciones de sensibilidad y drogorresistencia, existen reportes que indican que pueden variar con el estrés ambiental y mejorar el estado físico de MTB (22), lo que podría ayudar a entender mejor su rol en los aislados sensibles. Otras secuencias como transposasas, están involucradas en eventos de microevolución, lo que confiere cambios funcionales en las adaptaciones de la cepa a un determinado hospedero (23).
Nuestro reciente estudio del aislado sensible INS-SEN, indicó que los SNPs en la categoría COG tipo Q, I y L resultan ser más frecuentes, además de las secuencias codificantes de PPE y PE-PGRS. La importancia de las PPE radica en la asociación a la variación antigénica (24), mecanismo usado por parte de organismos para alterar sus proteínas de superficie con el fin de evadir la respuesta inmune. Entre tanto, las PE-PGRS están asociadas a la variación antigénica y la evasión inmune (25), estas proteínas ricas en secuencias GC repetidas, aumentan los eventos de recombinación y proporcionan variación génica. Las PPE y PE-PGRS tienen un importante rol en la virulencia de MTB (26), lo que manifiesta sus patrones polimórficos de los aislados sensible y drogorresistente. INS-SEN posee una ventaja y es que probablemente tenga más virulencia frente a aislados MDR o XDR (27). Se requieren más análisis para explorar los efectos de las mutaciones y su relación con la virulencia y la patogenicidad.
Los análisis de los genomas de MTB (MDR y XDR) contienen regiones de mutación diferentes a las relacionadas con resistencia a fármacos que resultan interesantes en analizar, ya que pueden desempeñar un papel importante en la supervivencia, adaptación y su difusión. En este estudio, la cepa INS-MDR, de linaje LAM es resistente a RIF e INH, el cual presenta una alta similaridad con KZN1435, lo que evidencia una posible expansión de la familia KZN en nuestro país (16) e INS-XDR drogorresistente a RIF, INH, PZA, CAP, KAN y con alta similaridad a KZN 605 (17). Estos hallazgos indican que INS-SEN y INS-XDR se encuentran estrechamente relacionados por evidencia filogenética que las ubican en el mismo lado con mínima distancia y los análisis comparativos de alta similaridad de SNPs entre ellas, con tan solo 51 nucleótidos diferentes, frente a un total de más de 4 millones de nucleótidos en cada genoma.
Los análisis en conjunto de cepas MDR y XDR determinan qué metiltransferasas se encuentran con más variabilidad, y esto debido probablemente a su rol en la reparación del ADN de MTB lo que provee una estabilidad genética, a pesar de la exposición del entorno hostil de macrófagos (28), además de actuar como activador de drogas, por ejemplo a tiacetazona (29). También el hecho de que genes involucrados en la síntesis de riboflavina, cobalamina, glutamina, molibdeno, nicotidamina y magnesio chelatasa se encuentren exclusivamente en las MDR y XDR, lleva a considerar que MTB y su cambio de fitness (eficacia biológica) presionan que existan modificaciones o selección dentro de las drogorresistentes para sintetizar moléculas clave en su funcionamiento normal o la modificación de alternativas en sus rutas metabólicas (30). Mayores análisis son necesarios para determinar realmente la importancia de los SNPs en los aislados drogorresistentes, estos análisis nos acercarán más a lo que sucede en la dinámica del genoma de MTB drogorresistentes en los aislados peruanos.
En conclusión, los aislados peruanos identificados de MTB, de la familia LAM con sensibilidad a drogas y drogorresistentes (MDR y XDR) no se encuentran estrechamente relacionados. Además, que INS-SEN se encuentra relacionado a un brote F11 con 99,9% de similitud, mientras las cepas drogorresistentes INS-MDR y INS-XDR se encuentran asociadas al brote KZN de Sudáfrica. Identificamos SNPs presentes específicos de genomas sensible y para las secuencias genómicas drogorresistentes asociadas con virulencia y patogenicidad.
Agradecimientos: al Dr. Cesar Cabezas, exjefe del Instituto Nacional de Salud de Perú, quien durante su gestión apoyó para poder secuenciar las tres cepas peruanas. A la Dra. Lely Solari quien siendo Directora del Centro Nacional de Salud Pública nos apoyó en la realización de los análisis de las cepas. A Harrison Montejo del LBBM por su apoyo logístico.
Contribuciones de autoría: DT, MG, han participado en la concepción y diseño del artículo. Los procedimientos y resultados fueron realizados por DT, MG, los análisis y discusiones fueron realizados por DT, MG y KSL. La redacción del artículo estuvo a cargo DT, MG, KSL, HG. La revisión crítica la realizo HG. La versión final estuvo a cargo de HG.
Fuentes de financiamiento: este estudio fue financiado por el Centro Nacional de Salud Pública del Instituto Nacional de Salud de Perú.
Conflictos de interés: los autores declaran no tener conflictos de interés.
REFERENCIAS BIBLIOGRÁFICAS
1. World Health Organization. Global Tuberculosis Report 2014. Geneva: WHO; 2014. [ Links ]
2. Murray CJ, Ortblad KF, Guinovart C, Lim SS, Wolock TM, Roberts DA, et al. Global, regional, and national incidence and mortality for HIV, tuberculosis, and malaria during 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384(9947):1005-70. doi: 10.1016/S0140-6736(14)60844-8. [ Links ]
3. Caceres O, Rastogi N, Bartra C, Couvin D, Galarza M, Asencios L, et al. Characterization of the genetic diversity of extensively-drug resistant Mycobacterium tuberculosis clinical isolates from pulmonary tuberculosis patients in Peru. PloS One. 2014;9(12):e112789. doi: 10.1371/journal.pone.0112789. [ Links ]
4. Garaycochea O, Ticona E. Rutas de transporte público y situación de la tuberculosis en Lima, Perú. Rev Peru Med Exp Salud Publica. 2015;32(1):93-7. [ Links ]
5. Somoskovi A, Parsons LM, Salfinger M. The molecular basis of resistance to isoniazid, rifampin, and pyrazinamide in Mycobacterium tuberculosis. Respir Res. 2001;2(3):164-8. [ Links ]
6. Ford CB, Shah RR, Maeda MK, Gagneux S, Murray MB, Cohen T, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. 2013;45(7):784-90. doi: 10.1038/ng.2656. [ Links ]
7. Victor TC, de Haas PE, Jordaan AM, van der Spuy GD, Richardson M, Van Soolingen D, et al. Molecular characteristics and global spread of Mycobacterium tuberculosis with a western cape F11 genotype. J Clin Microbiol. 2004;42(2):769-72. [ Links ]
8. Ioerger TR, Koo S, No EG, Chen X, Larsen MH, Jacobs WR Jr., et al. Genome analysis of multi- and extensively-drug-resistant tuberculosis from KwaZulu-Natal, South Africa. PLoS One. 2009;4(11):e7778. doi: 10.1371/journal.pone.0007778. [ Links ]
9. Culqui DR, Trujillo OV, Cueva N, Aylas R, Salaverry O, Bonilla C. Tuberculosis en la población indígena del Perú 2008. Rev Peru Med Exp Salud Publica. 2010;27(1):8-15. [ Links ]
10. Iwamoto T, Grandjean L, Arikawa K, Nakanishi N, Caviedes L, Coronel J, et al. Genetic diversity and transmission characteristics of Beijing family strains of Mycobacterium tuberculosis in Peru. PloS One. 2012;7(11):e49651. doi: 10.1371/journal.pone.0049651. [ Links ]
11. Garcia de Viedma D, Chaves F, Inigo J. New route of importation of Mycobacterium tuberculosis Beijing genotype. Emerg Infect Dis. 2006;12(1):169-70. [ Links ]
12. Taype CA, Agapito JC, Accinelli RA, Espinoza JR, Godreuil S, Goodman SJ, et al. Genetic diversity, population structure and drug resistance of Mycobacterium tuberculosis in Peru. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases. Infect Genet Evol. 2012;12(3):577-85. doi: 10.1016/j.meegid.2012.02.002. [ Links ]
13. Sheen P, Couvin D, Grandjean L, Zimic M, Dominguez M, Luna G, et al. Genetic diversity of Mycobacterium tuberculosis in Peru and exploration of phylogenetic associations with drug resistance. PloS One. 2013;8(6):e65873. doi: 10.1371/journal.pone.0065873. [ Links ]
14. Grandjean L, Iwamoto T, Lithgow A, Gilman RH, Arikawa K, Nakanishi N, et al. The Association between Mycobacterium Tuberculosis Genotype and Drug Resistance in Peru. PloS One. 2015;10(5):e0126271. doi: 10.1371/journal.pone.0126271. [ Links ]
15. Tarazona D, Borda V, Galarza M, Agapito JC, Guio H. Functional Analysis Using Whole-Genome Sequencing of a Drug-Sensitive Mycobacterium tuberculosis Strain from Peru. Genome Announc. 2014;2(1). pii: e00087-14. doi: 10.1128/genomeA.00087-14. [ Links ]
16. Galarza M, Tarazona D, Borda V, Agapito JC, Guio H. Evidence of Clonal Expansion in the Genome of a Multidrug-Resistant Mycobacterium tuberculosis Clinical Isolate from Peru. Genome Announc. 2014;2(1). pii: e00089-14. doi: 10.1128/genomeA.00089-14. [ Links ]
17. Guio H, Tarazona D, Galarza M, Borda V, Curitomay R. Genome analysis of 17 extensively drug-resistant strains reveals new potential mutations for resistance. Genome Announc. 2014;2(4). pii: e00759-14. doi: 10.1128/genomeA.00759-14. [ Links ]
18. Stavrum R, Valvatne H, Stavrum AK, Riley LW, Ulvestad E, Jonassen I, et al. Mycobacterium tuberculosis Mce1 protein complex initiates rapid induction of transcription of genes involved in substrate trafficking. Genes immun. 2012;13(6):496-502. doi: 10.1038/gene.2012.24. Epub 2012 Jun 14. [ Links ]
19. Chandolia A, Rathor N, Sharma M, Saini NK, Sinha R, Malhotra P, et al. Functional analysis of mce4A gene of Mycobacterium tuberculosis H37Rv using antisense approach. Microbiol Res. 2014;169(9-10):780-7. doi: 10.1016/j.micres.2013.12.008. [ Links ]
20. Senaratne RH, Sidders B, Sequeira P, Saunders G, Dunphy K, Marjanovic O, et al. Mycobacterium tuberculosis strains disrupted in mce3 and mce4 operons are attenuated in mice. J Med Microbiol. 2008;57(Pt 2):164-70. doi: 10.1099/jmm.0.47454-0. [ Links ]
21. Bibb LA, Hatfull GF. Integration and excision of the Mycobacterium tuberculosis prophage-like element, phiRv1. Mol Microbiol. 2002;45(6):1515-26. [ Links ]
22. Fan X, Abd Alla AA, Xie J. Distribution and function of prophage phiRv1 and phiRv2 among Mycobacterium tuberculosis complex. J Biomol Struct Dyn. 2016;34(2):233-8. doi: 10.1080/07391102.2015.1022602. [ Links ]
23. Perez-Lago L, Navarro Y, Herranz M, Bouza E, Garcia-de-Viedma D. Genetic features shared by Mycobacterium tuberculosis strains involved in microevolution events. Infect Genet Evol. 2013;16:326-9. doi: 10.1016/j.meegid.2013.02.016. [ Links ]
24. Zheng H, Lu L, Wang B, Pu S, Zhang X, Zhu G, et al. Genetic basis of virulence attenuation revealed by comparative genomic analysis of Mycobacterium tuberculosis strain H37Ra versus H37Rv. PloS One. 2008;3(6):e2375. doi: 10.1371/journal.pone.0002375. [ Links ]
25. Koh KW, Soh SE, Seah GT. Strong antibody responses to Mycobacterium tuberculosis PE-PGRS62 protein are associated with latent and active tuberculosis. Infect Immun. 2009;77(8):3337-43. doi: 10.1128/IAI.01175-08. [ Links ]
26. Ahmed A, Das A, Mukhopadhyay S. Immunoregulatory functions and expression patterns of PE/PPE family members: Roles in pathogenicity and impact on anti-tuberculosis vaccine and drug design. IUBMB Life. 2015;67(6):414-27. doi: 10.1002/iub.1387. [ Links ]
27. Smith KL, Saini D, Bardarov S, Larsen M, Frothingham R, Gandhi NR, et al. Reduced virulence of an extensively drug-resistant outbreak strain of Mycobacterium tuberculosis in a murine model. PloS One. 2014;9(4):e94953. doi: 10.1371/journal.pone.0094953. [ Links ]
28. Miggiano R, Casazza V, Garavaglia S, Ciaramella M, Perugino G, Rizzi M, et al. Biochemical and structural studies of the Mycobacterium tuberculosis O6-methylguanine methyltransferase and mutated variants. J Bacteriol. 2013;195(12):2728-36. doi: 10.1128/JB.02298-12. [ Links ]
29. Rombouts Y, Brust B, Ojha AK, Maes E, Coddeville B, Elass-Rochard E, et al. Exposure of mycobacteria to cell wall-inhibitory drugs decreases production of arabinoglycerolipid related to Mycolyl-arabinogalactan-peptidoglycan metabolism. J Biol Chem. 2012;287(14):11060-9. doi: 10.1074/jbc.M111.327387. [ Links ]
30. Gopinath K, Venclovas C, Ioerger TR, Sacchettini JC, McKinney JD, Mizrahi V, et al. A vitamin B12 transporter in Mycobacterium tuberculosis. Open Biol. 2013;3(2):120175. doi: 10.1098/rsob.120175. [ Links ]
Correspondencia:
David Tarazona
Dirección: Av. Defensores del Morro 2268, Chorrillos. Lima, Perú.
Teléfono: (+511) 7480000 anexo 1424
Correo electrónico: ddtarazona@gmail.com
Recibido: 20/10/2015
Aprobado: 09/03/2016
