










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
Print version ISSN 1726-4634
Rev. perú. med. exp. salud publica vol.34 no.2 Lima Apr./Jun. 2017
http://dx.doi.org/10.17843/rpmesp.2017.342.2484
ARTÍCULO ORIGINAL
Epidermólisis bullosa en el Perú: estudio clínico y epidemiológico de pacientes atendidos en un hospital pediátrico de referencia nacional, 1993-2015
Epidermolysis bullosa in Peru: clinical and epidemiological study of patients treated in a national reference pediatric hospital, 1993-2015
Rosario Torres-Iberico1,a, Patricia Palomo-Luck1,b, Gilmer Torres-Ramos1,c, Roxana Lipa-Chancolla1,d
1 Instituto Nacional de Salud del Niño. Lima, Perú.
a Médico dermatólogo; b licenciada en Nutrición, especialista en Nutrición Clínica; c odontólogo, especialista en Odontopediatría; d médico patólogo
RESUMEN
Objetivos. Describir las características clínicas y epidemiológicas de los pacientes diagnosticados con epidermólisis bullosa (EB), en el Instituto Nacional de Salud del Niño (INSN) en Lima, Perú; centro de referencia nacional para esta enfermedad. Material y métodos. Estudio observacional, descriptivo y transversal. Se revisaron las historias clínicas y exámenes de laboratorio de los pacientes diagnosticados de EB atendidos en el INSN desde 1993 al 2015. Resultados. Fueron registrados 93 pacientes. La edad promedio fue de 7,9 ± 5,6 años; el 53,8% (n=50) fueron hombres. Las formas clínicas correspondieron a EB distrófica con 41 (44,1%) casos, EB simple con 39 (41,9%) casos, EB de la unión con 8 (8,6%) y al síndrome de Kindler con 4 (4,3%) casos. No se pudo identificar la forma clínica en un caso. Procedían de Lima y Callao 48 casos (51,6%) y 45 casos (48,4%) de otras provincias del país. Entre las manifestaciones extracutáneas se registraron compromiso gastrointestinal (44,1%), ocular (37,6%), odontogénico (87,1%), nutricional (79,6%), además de pseudosindactilia (16,1%). Se halló desnutrición crónica (71,6%), desnutrición aguda (17,6%) y anemia en (62,4%). La mortalidad correspondió a 6 casos (6,5%). Conclusiones. Se reportan 93 casos de EB en el INSN, la presentación clínica predominante fue la forma distrófica.
Palabras clave: Epidermólisis ampollosa; Epidermólisis ampollosa distrófica; Epidermólisis ampollosa de la unión; Epidermólisis ampollosa simple (fuente: DeCS BIREME).
ABSTRACT
Objectives. To describe the clinical and epidemiological characteristics of patients diagnosed with epidermolysis bullosa (EB) at the Instituto Nacional de Salud (INSN) in Lima, Peru; a National Reference Center for this disease. Materials and methods. Observational, descriptive and transversal study. We reviewed the clinical histories and laboratory tests of patients diagnosed with EB treated in INSN from 1993 to 2015. Results. 93 patients were registered. The average age was 7.9 ± 5.6 years; 53.8% (n = 50) were boys. Clinical forms corresponded to dystrophic EB with 41 (44.1%) cases, simple EB with 39 (41.9%), union EB cases with 8 (8.6%) and Kindler syndrome with 4 (4.3%) cases. The clinical form could not be identified in a case. A total of 48 cases (51.6%) came from Lima and Callao, and 45 cases (48.4%) from other provinces of the country. Extracutaneous manifestations involved gastrointestinal (44.1%), ocular (37.6%), odontogenic (87.1%), and nutritional (79.6%) involvement, as well as pseudosindactilia (16.1%). Chronic malnutrition (71.6%), acute malnutrition (17.6%) and anemia (62.4%) were found. Mortality corresponded to 6 cases (6.5%). Conclusions. 93 cases of EB were reported in INSN, the predominant clinical presentation was the dystrophic form.
Key words: Epidermolysis bullosa; Epidermolysis bullosa dystrophica; Epidermolysis bullosa, junctional; Epidermolysis bullosa simplex (source: MeSH NLM).
INTRODUCCIÓN
La epidermólisis bullosa (EB) comprende a un grupo heterogéneo de enfermedades mecanobulosas hereditarias que se caracterizan por la susceptibilidad de la piel y mucosas a separarse de los tejidos subyacentes ante el mínimo traumatismo o roce, provocando la formación de ampollas y erosiones. Estas alteraciones son causadas por alrededor de 1000 mutaciones ubicadas en cerca de 18 genes que codifican proteínas estructurales de la piel (1). Se clasifica en cuatro grandes grupos, de acuerdo a la proteína alterada y al nivel, dentro de la epidermis, en el cual se forma la ampolla: EB simple, EB de la unión, EB distrófica y síndrome de Kindler (1).
La afectación cutánea se caracteriza por la presencia de ampollas en piel y mucosas, mientras que la extracutánea incluye alteraciones oculares, odontogénicas, gastrointestinales y músculo esqueléticas, entre otras (6,7). Incluso, se ha encontrado que algunos tipos de EB están asociados con el desarrollo de carcinoma epidermoide, carcinoma basocelular y melanoma maligno; riesgo que se incrementaría con la edad, sobre todo a partir de la tercera década de vida, por lo que se recomienda un seguimiento cercano para su detección y manejo temprano (8).
El compromiso de la salud de los afectados con EB rebasa el campo dermatológico, pues se presentan una gran variedad de manifestaciones sistémicas, psicológicas y sociales, mucho más graves que la propia enfermedad cutánea y que afectan notablemente la calidad de vida del paciente, por lo que se requiere un diagnóstico y manejo integral tempranos (4, 5).
La EB se ubica dentro de las enfermedades raras o huérfanas. Presenta una prevalencia de 8 casos por millón de habitantes en EE.UU., 10 casos por millón en Australia y 15-32 casos por millón en Inglaterra (2). En Latinoamérica, se registran 10,8 casos por millón de habitantes en Chile y 3,8 casos por millón en Uruguay (3). En el Perú, existen reportes aislados de pacientes adultos con EB pruriginosa (9); sin embargo, no hay estudios epidemiológicos que evalúen la situación de los niños con EB.
El objetivo de este estudio fue identificar las características clínicas y epidemiológicas de los pacientes diagnosticados con EB en el Instituto Nacional de Salud del Niño (INSN), centro de referencia nacional, entre los años 1993 – 2015; así mismo, tipificar las formas clínicas de EB y sus complicaciones más frecuentes.
MÉTODOS
TIPO DE ESTUDIO
Se realizó un estudio observacional, retrospectivo, transversal.
POBLACIÓN DE MUESTRA
Se revisaron las historias clínicas de pacientes diagnosticados de EB que fueron atendidos en el INSN desde 1993 al 2015.
CRITERIOS DE SELECCIÓN
Se incluyeron las historias clínicas de los pacientes con EB diagnosticados clínicamente y por estudio de microscopía óptica y/o electrónica de transmisión (MET) y/o mapeo antigénico por inmunofluorescencia (MAI), atendidos en el INSN durante el periodo comprendido entre los años 1993 y 2015. Se excluyeron aquellas historias clínicas que inicialmente tenían el diagnóstico de EB, pero que en su seguimiento fueron confirmadas como enfermedades ampollares de otra etiología, tales como herpes congénito neonatal, varicela congénita, entre otras.
VARIABLES
Se consideró a la clasificación recomendada por el Comité de Expertos del 2014 (1) para definir los tipos de EB, el tipo de presentación clínica (características cutáneas y extracutáneas) y los últimos exámenes auxiliares de control disponibles (hemoglobina, hierro sérico, transferrina, plaquetas, proteína C-reactiva [PCR], velocidad de sedimentación globular [VSG]), proteínas totales y fraccionadas, sedimento de orina). El estado nutricional fue evaluado en base a parámetros de peso y talla, de acuerdo a las tablas de la Organización Mundial de la Salud, versión 2008 (10). Para la severidad de la anemia se consideró a lo indicado en el instrumento iscorEB (en español: Instrumento para calificar los resultados para la investigación de la EB): anemia leve de 10 a 11,9 g/dL, anemia moderada de 8,0 a 9,9 g/dL y anemia severa de 6,1 a 7,9 g/dL (11).
Además, se incluyeron edad, sexo, procedencia (Lima-Callao, otras provincias), cobertura de salud (Seguro Integral de Salud [SIS], Seguro Social de Salud [EsSalud], Fondo de Salud para la Policía Nacional [FOSPOLI], y seguro privado) y mortalidad, que fue determinada de acuerdo al registro estadístico del INSN.
PROCEDIMIENTO
Inicialmente, se realizó la búsqueda de casos en el cuaderno de registro del Servicio de Dermatología y de los archivos estadísticos del INSN. Con la lista de casos identificados, se hizo la búsqueda de historias clínicas.
Se realizó el llenado de las fichas de recolección de datos de las historias clínicas desde 1993 hasta 2015 del INSN, posterior a la confirmación del diagnóstico. A continuación, se ingresaron los datos a una base electrónica en programas de Microsoft Excel y Word, versión para Windows 2007. Finalmente, se incluyeron los datos respecto a la mortalidad según el registro estadístico del INSN y, en 1 caso, por el reporte del médico del centro de salud.
ANÁLISIS ESTADÍSTICO
Se elaboró una base de datos en el programa Microsoft Excel (versión 2007, Redmond, EE. UU.) con los datos obtenidos. Se procedió al análisis estadístico expresado en tablas y gráficos según los tipos de variables. Las variables cualitativas fueron presentadas de acuerdo a su frecuencia (porcentajes), las variables cuantitativas en función de la media y su desviación estándar. Para dicho proceso se utilizó el SPSS, versión 15.0 (SPSS inc., Chicago, EE. UU.).
ASPECTOS ÉTICOS
El estudio fue presentado y aprobado por el Comité de Ética Institucional del INSN. Los datos fueron registrados y recodificados de tal forma que se mantuvo la confidencialidad de los mismos.
RESULTADOS
Del total de pacientes identificados, se excluyeron a 8 debido a que no se pudo hallar sus historias clínicas. Finalmente, se incluyeron a 93 pacientes con diagnóstico de EB en el estudio. La edad promedio fue de 7,9 ± 5,6 años, el 53,8% fueron hombres, 48 (51,6%) procedían de Lima y Callao, y 45 (48,4%) de otras provincias del país.
La frecuencia de EB en el grupo de 0 a 4 años fue de 32,3%, en el de 5 a 9 años fue 25,8%, en el de 10 a 14 años de 28,0%, en el de 15 a 19 años de 9,7% y en los mayores de 20 años fue de 4,3%. En cuanto a las formas clínicas, el 44,1% de los casos correspondió a EB distrófica, 41,9% a EB simple, 8,6% a EB de la unión, 4,3% a síndrome de Kindler y uno (1,1%) fue indeterminado.
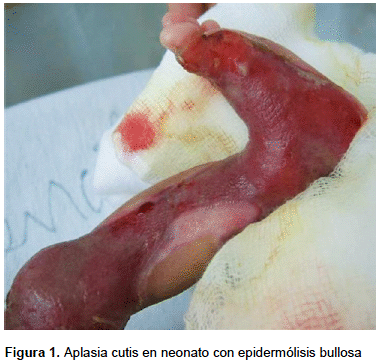
Además del clásico compromiso cutáneo de EB (ampollas), también se registraron 18 (19,4%) casos con aplasia cutis (Figura 1) y 4 (4,3%) con nevus EB.
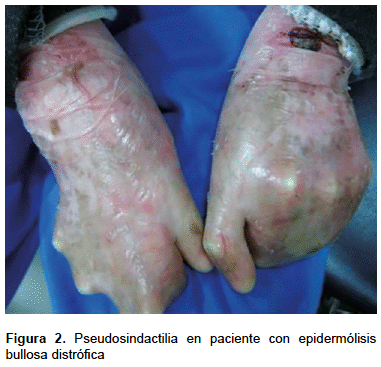
Dentro de las manifestaciones extracutáneas, hubo compromiso gastrointestinal en 41 pacientes (44,1%), oftalmológico en 35 (37,6%), odontogénico en 81 (87,1%), nutricional en 74 (79,6%) y renal y urológico en 6 (6,5%). El compromiso músculo esquelético correspondió en su totalidad a la pseudosindactilia con 15 casos (16,1%), todos ellos con EB distrófica (Figura 2). La anemia afectó a 58 (62,4%) pacientes, correspondiendo, en su mayoría, a pacientes con EB distrófica, seguida de las formas simple y de la unión (Tabla 1). El paciente con EB indeterminada no tuvo manifestaciones extracutáneas.
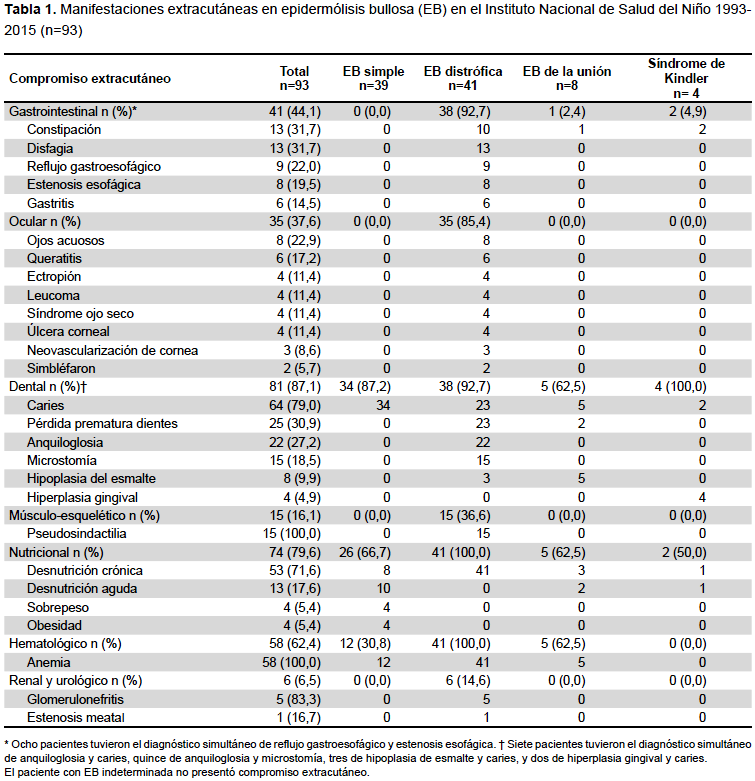
En el caso del compromiso renal y urológico, 5 pacientes tuvieron glomerulonefritis y 1 estenosis meatal; todos ellos tenían la forma clínica EB distrófica. En un caso severo se pudo identificar a la glomerulonefritis como infecciosa, debido a Estreptococcus b hemolítico del grupo A de Lancefield; en los demás casos solo se encontró hematuria, que incluso fue macroscópica en uno de ellos.
Respecto al compromiso gastrointestinal, las molestias más frecuentes fueron constipación (31,7%), disfagia (31,7%) y reflujo gastroesofágico (22,0%). La EB distrófica fue la que presentó más molestias gastrointestinales con 38 casos (92,7%).
El compromiso oftalmológico afectó solo a los pacientes con EB distrófica y las patologías más frecuentes incluyeron ojos acuosos (22,9%) y queratitis (17,2%), la menos frecuente simbléfaron (5,7%).
En el plano odontogénico, la caries fue la más frecuente (79,0%), seguido de pérdida dental prematura (30,9%) y la anquiloglosia (27,2%). El 35,9% de los casos con caries correspondieron a EB distrófica. La anquiloglosia solo se presentó en la EB distrófica.
Respecto al estado nutricional, se identificó desnutrición crónica (71,6%), desnutrición aguda (17,6%), sobrepeso (5,4%) y obesidad (5,4%). La desnutrición crónica estuvo presente en todas las formas clínicas, la desnutrición aguda se identificó en todas las formas, excepto EB distrófica, tanto el sobrepeso como la obesidad se presentaron solo en la forma clínica EB simple, con 4 casos cada uno.
Los niveles más bajos de hemoglobina y bajas reservas de hierro se registraron en las formas clínicas más severas de EB distrófica y EB de la unión. Asimismo, se evidenciaron niveles altos de PCR, VSG y plaquetas en las formas severas de EB distrófica. En 12 historias clínicas no se encontraron resultados de laboratorio que incluyeran hemoglobina, hematocrito, peso o talla para concluir si había anemia, desnutrición o retardo del crecimiento.
En las complicaciones, se registraron 10 casos (13,1%) de sepsis con punto de partida de piel y del tracto respiratorio. La mortalidad registrada fue de 6,5% debido a sepsis en 4 casos (2 de EB distrófica y 2 de EB de la unión) y 2 por insuficiencia respiratoria aguda en EB de la unión. El seguro de salud al cual estaban afiliados los pacientes SIS fueron 44 (47,3%); 41 (44,1%) pertenecían a EsSalud, 1 (1,1%) al FOSPOLI y 7 (7,5%) a seguro privado.
DISCUSIÓN
La forma más frecuente de EB fue la distrófica. Esto podría deberse a que esta forma clínica es la que presenta mayores complicaciones y, por tanto, mayores ingresos a unidades especializadas como el INSN. No obstante, ha habido un mayor número de casos con EB simple en los últimos 5 años, forma clínica que es la más frecuente en reportes de otros países (2); cabe la posibilidad, inclusive, de un subregistro, ya que los pacientes con EB simple no suelen presentar grandes cambios en su calidad de vida, por lo que no son derivados al INSN o no son diagnosticados adecuadamente.
Es probable que el registro de nuestro estudio refleje la casuística nacional. Primero, porque corresponde dentro del sistema nacional de salud a un hospital de nivel de atención III-2, es decir, un centro de referencia nacional en la atención pediátrica (12). Segundo, porque desde hace 22 años viene brindando atención a pacientes con EB procedentes de todo el país, y con diferentes tipos de seguro de salud
En cuanto al compromiso cutáneo, la aplasia cutis estuvo presente en 18 casos, tanto en la forma clínica de EB simple como EB de la unión y EB distrófica, coincidente con lo reportado en la literatura que indica que es más frecuente en estas dos formas (2). A inicios del año 1995, la aplasia cutis se trataba mediante el uso de membranas amnióticas y xenoinjerto; sin embargo, en los últimos 5 años se vienen utilizando apósitos de hidrofibra e hidrofílicos (13), con los que se ha logrado una rápida recuperación y reducción del dolor, especialmente en el neonato.
Se registraron 4 casos de nevus EB presentes en EB simple, EB de la unión y EB distrófica; usualmente, esta lesión melanocítica sigue un curso benigno, no obstante, puede tener manifestaciones clínicas, histopatológicas y dermatoscópicas sugestivas de melanoma, por lo que se recomienda su seguimiento (14).
No se registraron casos de malignidad por carcinoma epidermoide, carcinoma basocelular o melanoma maligno; probablemente porque estas neoplasias se presentan con más frecuencia entre la segunda y tercera década de la vida (8).
El compromiso extracutáneo en la EB causa una alta morbilidad y mortalidad (15). En nuestro estudio se presentaron casos con disfagia como manifestación del compromiso esofágico a muy temprana edad, seguido de estenosis y/o reflujo gastroesofágico. Si bien esta puede presentarse en todas las formas clínicas, estuvo presente con mayor severidad en la forma EB distrófica. Asimismo, la constipación se intensificó con la dificultad para masticar, deglutir y la administración de suplemento de fierro oral, similar a lo reportado (3).
El compromiso ocular estuvo presente en las formas de EB de la unión y EB distrófica, se llegaron a presentar, incluso, casos de vascularización corneal periférica y queratitis severa, tal como se reporta en otros estudios (16,17), problemas oftálmicos que requieren un manejo especializado.
El compromiso de cavidad oral incluyó lesiones en tejidos blandos y dientes. En la forma EB distrófica, la cicatrización y pérdida de la arquitectura normal de los tejidos blandos causó microstomía y anquiloglosia. En los casos de EB de la unión y distrófica, se presentó hipoplasia del esmalte, que junto a la pobre higiene oral, contribuyó a la pérdida prematura de dientes.
Dentro de las complicaciones por caries, se registraron pulpitis y necrosis pulpar, al igual que otros estudios (18-20). El servicio de odontopediatría del INSN brinda tratamientos preventivos y rehabilitadores, como coronas acrílicas temporales, que favorecieron la buena masticación y una mejor nutrición en los niños. Sin embargo, la desnutrición crónica fue frecuente, en especial en los pacientes con EB distrófica. Esto, probablemente, haya sido consecuencia de la disfagia, microstomía y pérdida de nutrientes por las erosiones en piel y mucosas, llegándose a registrar un retardo en el crecimiento hasta un déficit menor a 2 desviaciones estándar en los púberes.
Se ha reportado que el estado hipercatabólico y la inflamación crónica existentes en las formas severas de EB, producto de la liberación de citoquinas proinflamatorias (interleuquinas 1 y 6, factor de necrosis tisular) inhiben la eritropoyesis afectando negativamente el uso del hierro circulante, lo que conlleva a retardo del crecimiento (21,22). Más del 60% de pacientes con EB presentaron anemia, cifra que podría ser mayor ya que el 12,9% de los casos no tenía estudios de laboratorio; además, 45 pacientes procedían de provincia, lo que dificultó su seguimiento clínico y de laboratorio. Por todo ello, son importantes el consejo y soporte nutricionales personalizados para promover su crecimiento y desarrollo, optimizar el cierre de las heridas y mejorar su calidad de vida (23).
El 16,1% de los casos –todos con EB distrófica- presentó pseudosindactilia ya sea en manos o pies, alteración que es producto de la fibrosis y cicatrización de heridas (24). Esta se inicia a muy temprana edad con la aducción del primer espacio interdigital y luego contracturas en flexión con limitación funcional del movimiento, por lo que requieren tratamientos quirúrgicos y de rehabilitación para mejorar el deterioro funcional (24). Se sugiere que esta fibrosis es mediada por el factor de crecimiento transformante beta (25). Actualmente, la terapia está dirigida a reducir este proceso. Estudios en ratones hipomórficos con EB distrófica, tratados con losartán (antagonista del receptor tipo 1 de la angiotensina II) han mostrado reducir la fibrosis e inflamación (25). Otras drogas en estudio incluyen el ruxolitinib, pirfenidone y nintedanib (25).
En cuanto al compromiso renal, pudimos identificar un caso con glomerulonefritis posinfecciosa por Estreptococcus b hemolítico del grupo A de Lancefield e, incluso, hematuria macroscópica. Se ha reportado que las causas de glomerulonefritis en casos con EB corresponden a nefropatia por IgA, amiloidosis renal, nefritis intersticial, glomerulonifritis posinfecciosa e insuficiencia renal crónica (26). El compromiso renal ocupa el segundo lugar de causa de muerte en EB después del carcinoma epidermoide (26). Así mismo, se encontró estenosis del meato urinario en uno de nuestros casos, similar a lo reportado en la literatura (27). Por lo tanto, es necesario monitorizar la función renal, incluyendo uroanálisis, y la presión arterial en estos pacientes, a fin de una detección temprana y así reducir el riesgo de complicaciones posteriores.
La sepsis con punto de partida en piel (en EB distrófica) y en pulmón (en EB de la unión) fue la causa principal de mortalidad, coincidiendo con lo reportado en la literatura (15).
Actualmente existen mejoras en la terapéutica de EB tales como la terapia génica, trasplante de células pluripotenciales (fibroblastos alogénicos, células madre mesenquimales, médula ósea alogénicas) y remplazo proteico (28). Sin embargo, esta enfermedad sigue siendo de evolución crónica, llegando a mermar la calidad de vida de los pacientes y su supervivencia, lo cual supone un reto tanto para los afectados como para los profesionales que los atienden.
En nuestro medio existe poco conocimiento de la enfermedad por parte del personal de salud y se carece de protocolos de manejo. Además, existe dificultad para disponer de material de curación debido a su elevado costo y por ser usado diariamente. En respuesta, se aprobó según RD 354-2015-INSN- DG en el INSN el Programa "Atención Sanitaria, Social y Psicológica para pacientes con Epidermólisis Bullosa" y se conformó el Equipo Multidisciplinario de Atención que incluye la participación de dermatología, nutrición, odontología, anatomía patológica, fisioterapia, psicología, asistencia social, farmacia y comunicaciones para la atención integral del paciente con EB.
A nivel gubernamental, se requiere la aprobación de documentos normativos tales como el reglamento de la Ley 29 698 "Ley que Declara de Interés Nacional y Preferente Atención el Tratamiento de Personas que Padecen Enfermedades Raras o Huérfanas" del año 2011 o el "Plan Nacional de las Enfermedades Huérfanas 2016-2021" cuyo fin es garantizar el derecho al acceso a los servicios de salud, así como la gestión de promoción, prevención, diagnóstico, tratamiento integral y rehabilitación, en un marco de seguridad del paciente y financiamiento con cargo a los recursos asignados al Fondo Intangible Solidario de Salud. Actualmente, su cobertura es brindada de forma parcial por el SIS, FOSPOLIS y EsSalud.
Dentro de las limitaciones del estudio, se identificó que pudiera haber un subregistro de síntomas y complicaciones en el caso de los pacientes de provincia; algunos acudieron inicialmente, pero no se realizaron más evaluaciones de seguimiento y es probable que no se hayan registrado todas las posibles manifestaciones cutáneas, extracutáneas, complicaciones y mortalidad.
A pesar de que el diagnóstico de EB fue realizado mediante la clínica y estudio de microscopía electrónica e inmunomapeo, es necesario que la evaluación incluya un estudio genético (secuenciación génica completa) para determinar el patrón de herencia, el tipo de mutación e identificación los subtipos, que permitan establecer (29,30) e el pronóstico de la enfermedad implementar intervenciones sanitarias (asesoramiento genético, etc.).
Se concluye que la forma clínica predominante fue la EB distrófica. Las manifestaciones extracutáneas más frecuentes fueron desnutrición, anemia y caries dental.
La complicación más frecuente fue sepsis, cuyos focos fueron en piel y pulmón. La mayoría de pacientes tenían un seguro SIS o EsSalud. Se sugiere la implementación de un registro nacional de pacientes con EB con el fin de aplicar políticas de salud, y diseñar estrategias de comunicación para brindar información de la enfermedad con el fin de generar responsabilidad social y motivar la participación de la sociedad civil para enfrentar este problema.
Contribuciones de autoría: RTIP contribuyó con la concepción del estudio, el diseño y la redacción. Todos los autores contribuyeron con la recolección, análisis, interpretación y aprobación de la versión final del artículo.
Fuentes de financiamiento: autofinanciado.
Declaración de conflictos de interés: los autores declaran no tener conflictos de interés.
REFERENCIAS BIBLIOGRÁFICAS
1. Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, Has C, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70(6):1103-26. doi: 10.1016/j.jaad.2014.01.903. [ Links ]
2. Browne F, Heagerty A, Martinez A, Mellerio J, Moss. RF-8 The epidemiology of epidemolysis bullosa in the U.K.: a 9-year study. Br J Dermatol. 2011;165(Suppl. 1):11. doi: 10.1111/j.1365-2133.2011.10319.x. [ Links ]
3. Tabares F, Diaz C, Más V, Monteghirfo R. Epidermólisis ampollosa congénita: a propósito de un caso. Arch Med Inter. 2015;37(3):135-9.
4. Uitto J, Bruckner-Tuderman L, Christiano A, McGrath J, Has C, South A, Kopelan B, and E. Robinson E. Progress towards treatment and cure of epidermolysis bullosa: summary of the DEBRA International Research Symposium EB2015. J Invest Dermatol. 2016;136(2):352-8. doi: 10.1016/j.jid.2015.10.050. [ Links ]
5. Vanden Oever MJ, Tolar J. Advances in understanding and treating dystrophic epidermolysis bullosa. F1000Prime Rep. 2014;6:35. doi: 10.12703/P6-35. [ Links ]
6. Van der Kooi-Pol MM, Duipmans JC, Jonkman MF, van Dijl JM. Host pathogen interactions in epidermolysis bullosa patients colonized with Staphylococcus aureus. Int J Med Microbiol. 2014;304(2):195-203. doi: 10.1016/j.ijmm.2013.11.012. [ Links ]
7. Mellerio JE. Infection and colonization in epidermolysis bullosa. Dermatol Clin. 2010;28(2):267-9. doi: 10.1016/j.det.2010.01.004. [ Links ]
8. Mellerio JE, Robertson SJ, Bernardis C, Diem A, Fine JD, George R, et al. Management of cutaneous squamous cell carcinoma in patients with epidermolysis bullosa: best clinical practice guidelines. Br J Dermatol. 2016;174(1):56-67. doi: 10.1111/bjd.14104. [ Links ]
9. Ponce M, Paucar S, Mendoza R, Paredes G. Epidermólisis bulosa distrófica pruriginosa dominante. Folia dermatol Peru. 2010;21(1):45-9. [ Links ]
10. World Health Organization. WHO child growth standards: length/height for age, weight-for-age, weight-forlength, weight-for-height and body mass index-for-age, methods and development [Internet]. Ginebra: WHO; 2008 [citado el 10 de enero de 2017]. Disponible en: http://www.who.int/childgrowth/standards/technical_report/en. [ Links ]
11. Schwieger-Briel A, Chakkittakandiyil A, Lara-Corrales I, Aujla N, Lane AT, Lucky AW, et al. Instrument for scoring clinical outcome of research for epidermolysis bullosa: a consensus-generated clinical research tool. Pediatr Dermatol. 2015;32(1):41-52. doi: 10.1111/pde.12317. [ Links ]
12. Instituto Nacional de Salud del Niño. Boletín Informativo. Lima: Oficina Ejecutiva de Apoyo a la Investigación y Docencia Especializada; Junio 2016. Número 2.
13. El Hachem M, Zambruno G, Bourdon-Lanoy E, Ciasulli A, Buisson C, Hadj-Rabia S, et al. Multicentre consensus recommendations for skin care in inherited epidermolysis bullosa. Orphanet J Rare Dis. 2014;9:76. doi: 10.1186/1750-1172-9-76. [ Links ]
14. Lanschuetzer CM, Laimer M, Nischler E, Hintner H: Epidermolysis Bullosa Nevi. Dermatol Clin 2010;28(1):179-83. doi: 10.1016/j.det.2009.10.024. [ Links ]
15. Hon KL, Li JJ, Cheng BL, Luk DC, Murrell DF, Choi PC, Leung AK. Age and etiology of childhood epidermolysis bullosa mortality. J Dermatolog Treat. 2015;26(2):178-82. doi: 10.3109/09546634.2014.915002. [ Links ]
16. Figueira EC, Murrell DF, Coroneo MT. Ophthalmic involvement in inherited epidermolysis bullosa. Dermatol Clin. 2010;28(1):143-52. doi: 10.1016/j.det.2009.10.021. [ Links ]
17. Díaz M, Pérez Z, Escalona E, Noriega J, Castillo A, Márquez S. Manifestaciones oftalmológicas de la epidermólisis bullosa. Rev Cubana Oftalmol. 2014;27(4):619-25. [ Links ]
18. Kudva P, Jain R. Periodontal manifestation of epidermolysis bullosa: looking through the lens. J Indian Soc Periodontol. 2016;20(1):72-4. doi: 10.4103/0972-124X.164760. [ Links ]
19. Kramer SM, Serrano MC, Zillmann G, Gálvez P, Araya I, Yanine N, et al. Oral health care for patients with epidermolysis bullosa - best clinical practice guidelines. Int J Paediatr Dent. 2012;22(Suppl. 1):1-35. doi: 10.1111/j.1365-263X.2012.01247.x. [ Links ]
20. Valle M, Krämer S; Véliz A, Leiva N, Vergara C; Zillmann G. Técnica alternativa y simple de rehabilitación oral con prótesis removible para una paciente con epidermólisis bullosa distrófica: reporte de caso clínico. Rev Clin Periodoncia Implantol Rehabil Oral. 2015;8(3):244-8. doi: 10.1016/j.piro.2015.10.001. [ Links ]
21. Martinez A, Alllgrove J, Brian C. Growth and pubertal delay in patients with epidermolysis bullosa. Dermatol Clin. 2010;28(2):357-9. doi: 10.1016/j.det.2010.01.007. [ Links ]
22. Fu T; Lingala B; Kent K, Bachrach LK, Bruckner AL. Patterns of bone mineral acquisition in children with epidermolysis bullosa: a longitudinal study. Br J Dermatol. 2011;165(5):1081-6. doi: 10.1111/j.1365-2133.2011.10517.x. [ Links ]
23. Zidorio AP, Dutra ES, Leão DO, Costa IM. Nutritional aspects of children and adolescents with epidermolysis bullosa: literature review. An Bras Dermatol. 2015;90(2):217-23. doi: 10.1590/abd1806-4841.20153206. [ Links ]
24. Luria S, Radwan S, Zinger G, Eylon S. Hand Surgery for Dystrophic Epidermolysis Bullosa. J Pediatr Orthop. 2014;34(7):710-4. doi: 10.1097/BPO.0000000000000152. [ Links ]
25. Nyström A, Thriene K, Mittapalli V, Kern J, Kiritsi D, Dengjel J. Losartan ameliorates dystrophic epidermolysis bullosa anduncovers new disease mechanisms. EMBO Mol Med. 2015;7(9):1211-28. doi: 10.15252/emmm.201505061. [ Links ]
26. Harikrishnan KN, Krishnamurthy S, Rajesh NG, Mahadevan S . Renal involvement in epidermolysis bullosasimplex: an unusual presentation. Indian J Pediatr. 2014;81(1):102-3. doi: 10.1007/s12098-012-0907-5. [ Links ]
27. Almaani N, Mellerio JE. Genitourinary tract involvement in epidermolysisbullosa. Dermatol Clin. 2010;28(2):343-6. doi: 10.1016/j.det.2010.01.014. [ Links ]
28. Cohn HI, Teng JM. Advancement in management of epidermolysis bullosa. Curr Opin Pediatr 2016;28(4):507-16. doi: 10.1097/MOP.0000000000000380. [ Links ]
29. Berk DR, Jazayeri L, Marinkovich MP, Sundram UN, Bruckner AL. Diagnosing epidermolysis bullosa type and subtypein infancy using immunofluorescencemicroscopy: the Stanford experience. Pediatr Dermatol. 2013;30(2):226-33. doi: 10.1111/j.1525-1470.2012.01880.x. [ Links ]
30. Takeichi T, Liu L, Fong K, Ozoemena L, McMillan JR, Salam A, et al. Wholeexome sequencing improves mutationdetection in a diagnostic epidermolysisbullosa laboratory. Br J Dermatol. 2015;172(1):94-100. doi: 10.1111/bjd.13190. [ Links ]
Correspondencia: Rosario Torres Iberico
Dirección: Av. Belén 323 Dpto. 401, San Isidro. Lima, Perú.
Teléfono: (+511) 999289587
Correo electrónico: rtorresi@insn.gob.pe
Recibido: 07/11/2016
Aprobado: 24/05/2017
En línea: 28/06/2017

