










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
Print version ISSN 1726-4634
Rev. perú. med. exp. salud publica vol.35 no.3 Lima July/Set. 2018
http://dx.doi.org/10.17843/rpmesp.2018.353.3317
CARTAS AL EDITOR
Registro de pacientes con inmunodeficiencias primarias en los tres principales centros de referencia del Perú
Registration of patients with primary immunodeficiencies in the three main reference centers in Peru
David García-Gomero 1,2,3,a, Wilmer Córdova-Calderón1,3,4,b, Juan Aldave-Becerra 1,3,5,b
1 Centro de Referencia Nacional de Alergia, Asma e Inmunología. Instituto Nacional de Salud del Niño. Lima, Perú.
2 Facultad de Medicina. Universidad Nacional Mayor de San Marcos. Lima, Perú.
3 Sociedad Peruana de Inmunología. Lima, Perú.
4 Grupo peruano de Inmunodeficiencias Primarias (PERUGID). Lima, Perú.
5 Servicio de Alergias e Inmunología. Hospital Nacional Edgardo Rebagliati Martins. Lima, Perú.
a Médico cirujano, b Médico inmunólogo clínico-alergólogo.
Sr. Editor. Las inmunodeficiencias primarias (IDP) incluyen un conjunto de trastornos hereditarios de la función del sistema inmunitario (1). Según clasificación de la International Union of Immunological Societies (IUIS) estos trastornos se pueden dividir en nueve categorías: inmunodeficiencias combinadas, deficiencias predominantemente de anticuerpos, inmunodeficiencias combinadas con síndromes bien definidos, enfermedades de desregulación inmune, defectos congénitos del número, función o ambos del sistema fagocítico; defectos de la inmunidad innata, desordenes autoinflamatorios, deficiencias del complemento e inmunodeficiencias por fenocopias (2).
Existen signos de sospecha de IDP establecidos por la Jeffrey Modell Foundation (JMF) que se basan en la aparición de infecciones recurrentes en diferentes aparatos y sistemas (1). La Latin American Society for Immunodeficiencies (LASID) creó un sistema de registro para los casos de IDP (3). En el Perú, existen tres centros de referencia públicos que contribuyen con LASID en el registro de los pacientes con IDP: el Centro de Referencia Nacional de Alergia, Asma e Inmunología (CERNAAI) del Instituto Nacional de Salud del Niño (INSN), el Hospital Nacional Edgardo Rebagliati Martins (HNERM) y el Hospital Nacional Guillermo Almenara Irigoyen (HNGAI). Pretendemos realizar un reporte del número de casos y algunas características de los pacientes con IDP atendidos en estos centros.
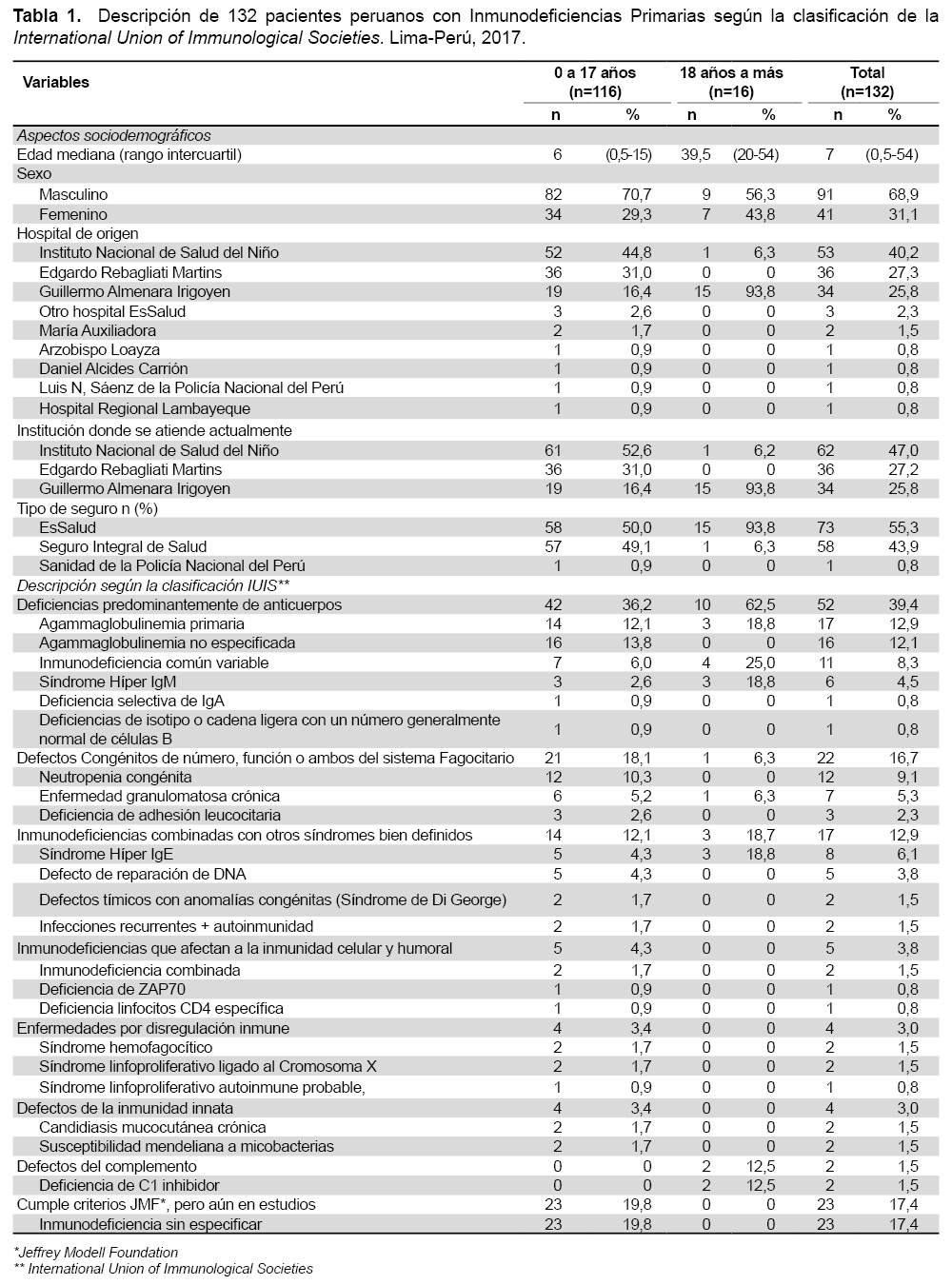
Se hizo revisión de los registros anónimos de los pacientes atendidos entre agosto del 2015 y abril del2017, previamente se obtuvo la autorización de las instancias pertinentes en cada centro. Se obtuvieron datos como edad, género, institución, tipo de seguro, diagnóstico y grupo al cual pertenece según IUIS. Se incluyeron a 116 pacientes menores de 18 años y 16con 18 años o más. La edad mediana fue 6(rango intercuartil: 0,05-15) años para los pacientes menores de 18 años y 47,5 (rango intercuartil:20-54) para los de 18 años o más; 91/132 (68%) fueron varones. Para las categorías de la IUIS, las deficiencias predominantemente de anticuerpos se hallan en 39,4 % (52/132) de los pacientes, de estos 32,7 %(17/52) corresponden a agammaglobulinemias primarias y 30,7 % (16/52) corresponden a agammaglobulinemias no especificadas.
La inmunodeficiencia común variable se halla en 21,2 % (11/132), los defectos congénitos del número, función o ambos del sistema fagocitario los encontramos en 16,7 % (22/132); entre ellas se encontró neutropenia congénita en 54,5 % (12/22), enfermedad granulomatosa crónica en 31,8 % y deficiencia de adhesión leucocitaria en 13,6% (3/22). Las inmunodeficiencias combinadas con otrossíndromes bien definidos fueron encontradas en 13,6 % (18/132) de los pacientes, entre ellos se identificaron el Síndrome Hiper IgE en 44,4 % (8/18), a la Ataxia Telangiectasia en 27,8 % (5/18) y al Síndrome de Di George en 11,1 % (2/18).
Las inmunodeficiencias que afectan la inmunidad celular y humoral afectaron al 3,0 % (4/132) de pacientes; las enfermedades por disregulación inmune afectaron al 3,0 % (4/132); al igual que los defectos de la inmunidad innata(3,0 %, 4/132) y los defectos del complemento afectado al 1,5% (2/132)pacientes. Finalmente encontramos que 16,7 % (22/132) pacientes cumplen con los criterios de la Jeffrey Model Foundation (JMF), pero aún se hallan en estudios. No encontramos pacientes con síndromes autoinflamatorios, ni Inmunodeficiencias por fenocopias. (Tabla 1).
Se puede concluir que la mayor parte de los pacientes acuden a establecimientos del seguro social (EsSalud) y que, tal como se describe en la literatura (5), los defectos en la producción de anticuerpos son la categoría predominante y, dentro de ellos, las agammaglobulinemias primarias. Por último, ya que la mayor parte de pacientes se halla en espera de un diagnóstico específico, se requiere implementar la logística necesaria en los centros hospitalarios para brindar un diagnóstico y tratamiento tempranos.
Fuentes de financiamiento: Grupo peruano de inmunodeficiencias primarias– PERUGID.
Conflictosde interés: Ninguno
Contribuciones de autoría: DGG y WCC han participado en la concepción del artículo. JAB y WC C participaron en la recolección de datos. DGG se encargó del procesamiento, análisis de datos y redacción del artículo. JAB y WC participaron en la revisión crítica del artículo y aprobación de la versión final. WCC obtuvo el financiamiento.
REFERENCIAS BIBLIOGRÁFICAS
1. PicardC, Al W, Bousfiha A, Casanova J, Chatila T, Conley M, et al. Primary Immunodeficiency Diseases: an Update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol. 2015;35(8):696-726. [ Links ]
2. Bousfiha A, Jeddane L, Al-Herz W, Ailal F, Casanova J, Chatila T, et al. The 2015 IUIS Phenotypic Classificationfor Primary Immunodeficiencies. J Clin Immunol. 2015;35(8):727-38. [ Links ]
3. Costa B, González M, Espinosa S, Segundo G. Latin American challenges with the diagnosis and treatment of primary immunodeficiency diseases. Expert Rev Clin Immunol. 2016; 21:1-7. [ Links ]
4. Modell V, Quinn J, Ginsberg G, Gladue R, Orange J. Jeffrey. Modell Foundation modeling strategy to identify patients with primary immunodeficiency utilizing risk management and outcome measurement. Immunol Res. 2017;65(3):713-20. [ Links ]
5. Bonilla F, Khan D, Ballas Z, Chinen J, Frank M, Hsu J, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015;136(5):1186-1205. [ Links ]
Correspondencia: David Santiago García Gomero.
Dirección: Calle Quipán 157 Urb. Tahuantinsuyo-Independencia.
Teléfono: 977325248
Correo electrónico: dsantg.g@gmail.com
Recibido: 21/12/2017
Aprobado: 17/01/2018
En línea: 29/08/2018

