











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
MENSAJES CLAVE
Motivación para realizar el estudio. El presente estudio se realizó por la dificultad que presenta el diagnóstico temprano y correcto de la distrofia muscular de Duchenne/Becker.
Principales hallazgos. Gracias a este estudio se ha logrado implementar una técnica nueva (MLPA) en el Perú para un mejor diagnóstico de la enfermedad.
Implicancias. Las distrofias musculares Duchenne/Becker son enfermedades raras, y es por su baja incidencia que no genera un gran interés, sin embargo, es importante implementar nuevas técnicas para el diagnóstico de las mismas, así como en su momento se implementó el diagnóstico de las enfermedades metabolómicas en los neonatos.
Todos los tipos de distrofia muscular son enfermedades que producen debilidad muscular, las más comunes son la distrofia muscular de Duchenne (DMD) y la de Becker (BMD) y son causadas por variaciones del gen DMD, uno de los más grandes del genoma humano (2.4Mb), compuesto por 79 exones y siete promotores, ubicado en la región Xp21.1 1. Dicho gen se expresa en músculo esquelético, cardiaco, células neuronales y músculo liso, y se traduce en distrofina, proteína que forma parte del complejo proteico asociado a distrofina (DAP), cuya función es estructural, pues debe estabilizar el sarcolema y proteger a las fibras musculares del daño y necrosis ocasionados por la contracción a largo plazo 1,2.
La DMD (OMIM 310200) es la más frecuente y tiene una incidencia de 1 por cada 3500 niños nacidos vivos 3. Tiene un pronóstico grave y expectativa de vida reducida. La BMD (OMIM 300376) tiene una incidencia de 1 por cada 30 000 niños nacidos vivos y tiene una forma más leve de presentación, con pronóstico de vida cercano a los 60 años 3.
Los casos de DMD son más graves debido a que la distrofina se presenta muy reducida, no funcional o en ausencia total, debido teóricamente a variaciones fuera del marco de lectura en el gen DMD3,4. Mientras que en los casos de DMB, las variaciones se encuentran dentro del marco de lectura, expresando una proteína incompleta pero funcional y por lo tanto con mejor pronóstico para el paciente 3,4.
En Perú, como en la mayoría de países en vías de desarrollo, el diagnóstico para DMD/DMB consta básicamente de la identificación de diferentes signos clínicos, como dificultad para caminar, hipotonía, atrofia muscular, signo de Gowers, escoliosis, contracturas articulares, y en cuadros avanzados, insuficiencia respiratoria y/o cardiaca; este diagnóstico se apoya en el nivel de creatina quinasa (CPK), elevada en varones si es mayor a 174 u/L; y en la electromiografía (EMG), que mide la actividad eléctrica de los músculos y distingue entre patrón neuromuscular normal, miopático o neuropático 4-7.
Como DMD/DMB son enfermedades monogénicas, todas las técnicas de diagnóstico confirmatorias están relacionadas a la detección directa de la distrofina in situ, como el análisis inmunohistoquímico o Western blot 4-7, y a la identificación de las variantes patológicas del mismo gen DMD, usando la técnica de reacción en cadena de la polimerasa multiplex (PCR-multiplex) (Test de Chamberlain, Beggs o Kunkel) que detecta sólo presencia/ausencia de unos 9 a 11 exones por reacción 5, o la técnica de amplificación múltiple dependiente de ligación por sondas (MLPA) que detecta la presencia/ausencia y dosis de los 79 exones y el promotor Dp427c en dos reacciones 8. Además, la secuenciación Sanger detecta pequeñas mutaciones en todas las regiones del gen DMD (una reacción por región) y la secuenciación de nueva generación (NGS) detecta la ausencia de grandes regiones (multiexónicas) y pequeñas mutaciones en todo el gen en una sola reacción 9,10.
El objetivo del presente estudio fue implementar la técnica MLPA para el diagnóstico de DMD/DMB y demostrar su ventaja sobre la PCR-multiplex, comúnmente utilizada en algunos centros hospitalarios del Perú.
EL ESTUDIO
El presente estudio es de tipo descriptivo prospectivo, comprende la primera parte de un proyecto de investigación denominado «Diagnóstico genético molecular en pacientes con Distrofia Muscular de Duchenne (DMD) y Becker (BMD) en Perú», que busca caracterizar genéticamente a 300 pacientes por diferentes técnicas moleculares durante tres años. El proyecto está financiado por la Facultad de Medicina Humana de la Universidad de San Martín de Porres.
Durante el primer año se reclutaron 60 pacientes provenientes del Servicio de Genética y Errores Innatos del Metabolismo del Hospital del Niño y del Servicio de Genética del Departamento de Especialidades Médicas del Hospital Edgardo Rebagliati Martins. Para este estudio se consideraron a 40 pacientes y a dos familias nucleares (de los únicos dos pacientes que aceptaron toma de muestra).
Además, se utilizaron diez controles masculinos sanos del banco de ADN del Centro de Investigación de Genética y Biología Molecular (CIGBM), tal como se recomienda en el protocolo de MLPA-DNA versión MDP-v003 de MRC-Holland 11.
Se incluyeron a pacientes peruanos, varones (por ser los afectados en una enfermedad ligada al X), menores de 18 años, diagnosticados clínicamente con distrofia muscular que presentan pruebas de apoyo al diagnóstico, CPK elevada y electromiografía que evidencia un patrón miopático. Se excluyeron pacientes con información clínica incompleta.
Se extrajo ADN (aproximadamente 500 uL) a partir de 3 mL de sangre periférica, siguiendo el protocolo descrito por Miller et al. («salting out») 12 estandarizado en el CIGBM.
Para la PCR-multiplex de nueve exones: exón 4 (186pb), exón 44 (268pb), exón 12 (331pb), exón 8 (360pb), exón 51 (328pb), exón 17 (417pb), exón 19 (459pb), exón 48 (505pb) y exón 45 (547pb) la amplificación se realizó según el protocolo descrito por Rojas, et al. 13. La detección mediante electroforesis utilizó agarosa de alta sensibilidad al 4%, con un programa de 30 voltios por 20 min, 60 voltios por 40 min, 90 voltios por una hora y 120 voltios por dos horas. La tinción fue con bromuro de etidio y las fotos se consiguieron con un fotodocumentador Biorad Gel Doc XR.
La MLPA consta de cuatro pasos: denaturación del ADN, hibridación con las sondas, ligación de estas y amplificación de los fragmentos que previamente han hibridado y ligado. Se realizó según el protocolo de MLPA-DNA versión MDP-v003 11, con pequeñas modificaciones (la hibridación duró de 16 a 20 horas y la ligación de las sondas duró 40 minutos). Se utilizaron los mixes de sondas comerciales: P034 y P035 (MRC-Holland), que en conjunto analizan los 79 exones del gen DMD y el promotor Dp427c. Los amplificados se corrieron por electroforesis capilar en un equipo ABI-3500, 1 uL de muestra más 9 uL de formamida HiDi y 0,2 uL de Liz 500. Se usó un capilar de 50 cm y POP 7, y las condiciones fueron de 1,6 KV de voltaje de inyección y 15 s de tiempo de inyección.
Siguiendo lo recomendado en el inserto de los mixes de sondas P034 y P035 se procedieron a confirmar las deleciones de un solo exón, mediante una reacción en cadena de polimerasa (PCR) de dos exones a la vez (el exón que tiene supuestamente la deleción y un exón control adicional).
Los datos extraídos de la electroforesis capilar se analizaron mediante el software libre Coffalyser.Net v.140721.1958 (www.mlpa.com), también recomendado por la MRC-Holland 11. Este análisis se realizó sobre el radio del DQ (dosaje de la fluorescencia captada por cada fragmento). Se evaluó la presencia o ausencia de cada fragmento y la dosis en que se presentan, en comparación con las muestras control.
Antes de la obtención de la muestra, los padres o apoderados de los participantes firmaron voluntariamente un consentimiento informado. El protocolo de investigación, así como el formato de consentimiento fue aprobado por el Comité Institucional de Ética en Investigación de la Universidad San Martín de Porres, el cual respeta las normas éticas concordantes con la declaración de Helsinki (actualizada al 2013).
HALLAZGOS
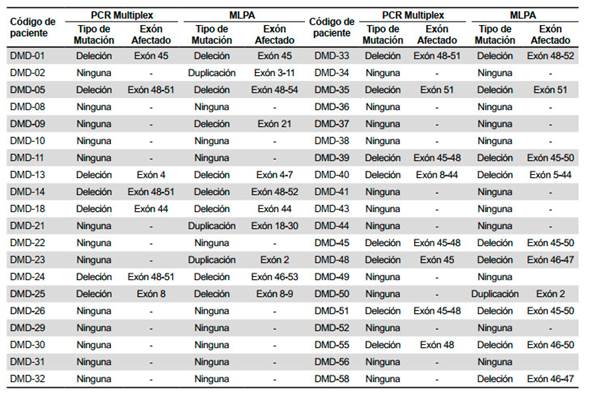
De las 40 muestras analizadas mediante PCR-multiplex, 15 (37,5%) presentaron deleciones de uno o más exones del gen DMD, causales de distrofia muscular (Tabla 1).
Tabla 1 Resultados de la técnica de reacción en cadena de la polimerasa multiplex (PCR multiplex) y de la técnica de amplificación múltiple dependiente de ligación por sondas (MLPA) en los 40 pacientes analizados
Estas deleciones se evidencian por la falta de las bandas correspondientes a determinados exones en comparación con el control (Figura 1). En las pruebas realizadas a las madres y/o hermanas de dos pacientes, no se encontró ninguna variación del patrón de bandas con respecto al control.
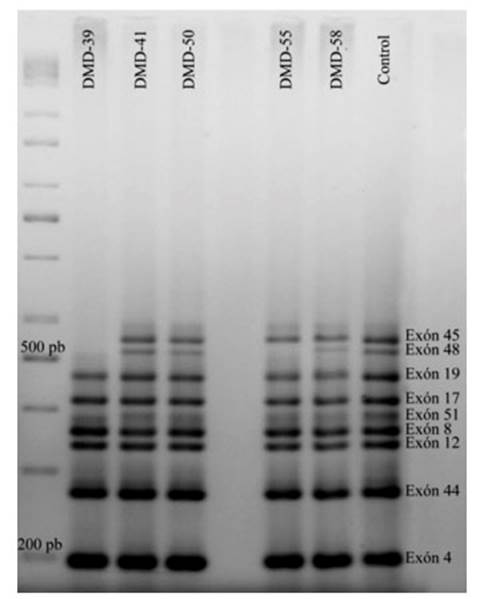
Figura 1 Electroforesis que muestra resultados de la PCR-multiplex; al lado izquierdo el marcador de 100-1000pb, al lado derecho un individuo control que presenta las nueve bandas esperadas correspondientes a los exones analizados por esta prueba. Los individuos DMD-41, DMD-50 y DMD-58 presentan las nueve bandas al igual que el control; en el individuo DMD-39 no se observan las bandas correspondientes a los exones 45 y 48, y en el individuo DMD-55 no está presente la banda correspondiente al exón 48
De las 40 muestras analizadas mediante MLPA, 21 (52,5%) fueron diagnosticados, de los cuales 17 (42,5%) corresponde a deleciones y 4 (10%) a duplicaciones de uno o más exones del gen DMD (Tabla 1). Además, las cuatro deleciones de un solo exón fueron verificadas por una PCR de dos exones. Al realizarse esta prueba a los familiares (madres y/o hermanas) de los mismos individuos escogidos anteriormente, estas resultaron ser portadoras.
En el pedigree de la familia A, mediante PCR-multiplex el paciente DMD-05 (II-1) tiene deleciones de los exones 48 y 51 del gen DMD (Figura 2), y mediante MLPA se determinó que la deleción realmente abarca desde el exón 48 hasta el 54. El paciente tiene un hermano menor (II-3) que también presenta la mutación confirmada por ambas técnicas, mientras que la hermana (II-2) y la madre (I-2) son portadoras de la mutación, detectadas sólo por MLPA (Figura 3).

Figura 2 Esquema de diagrama de cajas exportado del Coffalyser. A. Resultado de un individuo control (varón), se observa que para todos los exones el radio (DQ) se encuentra aproximadamente en uno (puntos negros) lo que indica que el paciente presenta el número de copias esperado de cada exón (una copia de una). B. Resultado del paciente DMD-05 (varón), se observa que para los exones del 48 al 54, el radio (DQ) es aproximadamente 0 (puntos rojos) lo que indica que el paciente presenta deleción de estos exones (ninguna copia de una). C. Resultado de la hermana del paciente DMD-05, se observa que para los exones del 48 al 54 el radio (DQ) es aproximadamente 0,5 (puntos rojos) lo que indica que la paciente es una portadora de la mutación (una copia de dos)
En el pedigree de la familia B, mediante PCR-multiplex el paciente DMD-18 (II-1) tiene una deleción del exón 44 del gen DMD y mediante la técnica MLPA se confirmó que sólo presenta la deleción de ese exón. Tiene dos hermanos menores (II-2 y II-3) que también presentan la misma mutación confirmada por ambas técnicas, mientras que la madre (I-2) es portadora de dicha mutación, detectada sólo por MLPA (Figura 3).

DISCUSIÓN
Actualmente en los centros hospitalarios del Perú, son poco recomendables las pruebas inmunohistoquímicas o de Western blot, ya que para ellas son necesarias muestras de biopsia muscular, consideradas muy invasivas. La PCRmultiplex se encuentra implementada desde hace varios años, la MLPA y el NGS no son de uso rutinario para el diagnóstico y la secuenciación Sanger se utiliza para el descarte de una mutación ya conocida, ya que implica un mayor gasto de tiempo y dinero 6,9,20.
En el presente estudio se demuestra que la MLPA diagnostica 15% más de casos que la PCR-multiplex; además, la MLPA confirma el 100% los casos detectados por PCR-multiplex y de estos un 80% se amplía y consigue un diagnóstico más exacto en cuanto al verdadero tamaño de la mutación. Estas ventajas permiten un mejor diagnóstico de la distrofinopatía que padece el paciente, y permite brindar un correcto pronóstico de la enfermedad. Estos resultados se corresponden con lo reportado por Schwartz & Duno 14, Lalic, et al. 15, Janssen, et al. 16, Lai, et al. 17 y Guo, et al. 18, quienes también confirmaron los resultados. Esto permite inferir que la técnica MLPA, para el análisis del gen DMD, es un método reproducible, fácil de manejar y más sensible que la PCR-multiplex.
Para citar un ejemplo, en el paciente DMD-24, la PCRmultiplex detecta la ausencia de los exones 48 y 51. Con MPLA se descubre que la verdadera mutación comprende una deleción desde el exón 46 hasta el exón 53. Esto es muy importante, ya que de darse el resultado de PCR-multiplex como una deleción del exón 48 al 51 no se altera el marco de lectura y por lo tanto se infiere que la proteína a formarse estaría alterada pero funcional, tratándose de un posible caso de DMB. Sin embargo, al conocerse que la verdadera mutación es desde el exón 46 hasta el 53 sí se altera el marco de lectura de la proteína parando tempranamente la traducción en el exón 45 por lo que se infiere una proteína muy pequeña que no presentaría su extremo terminal C. Esto evitaría la interacción de la distrofina con los miembros del DAP, ocasionando el cuadro clínico de Duchenne.

