











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La presencia de bacterias patógenas en el ambiente marino incrementa el interés humano en seguridad alimentaria debido a su potencial para causar brotes. Entre ellas, destaca la Vibrio parahaemolyticus, una bacteria gramnegativa halófila ampliamente distribuida en ecosistemas costeros, cuya serotipificación depende de los antígenos somáticos (O) y capsulares (K) producidos en diversas condiciones ambientales 1.
El interés por la V. parahaemolyticus se inició muchos años atrás, luego de ser agente causante de infecciones transmitidas por alimentos en un brote ocurrido en Japón. Históricamente, la V. parahaemolyticus ha sido responsable del 20-30% de los casos de infección alimentaria en Japón y otros países asiáticos 2. El Perú ha registrado brotes importantes desde 1997, los cuales han sido asociados a los cambios climáticos que forman parte del fenómeno de El Niño, y que alteran condiciones ecológicas marinas, como el incremento de la tasa de abundancia del plancton 3. La mayoría de estos informes asocian a la hemolisina directa termoestable (TDH, por sus siglas en inglés) con la virulencia de la V. parahaemolyticus 4.
Actualmente, la prevalencia mundial y la emergencia de casos de infección por V. parahaemolyticus van en incremento, subrayando la necesidad de mantener una vigilancia adecuada de este patógeno. La microbiología convencional resulta insuficiente para determinar variantes patógenas y su distribución geográfica. En contraste, herramientas de epidemiología molecular, como el esquema de tipificación multilocus (MLST, por sus siglas en inglés) se proponen como nuevas alternativas que permiten el estudio de enfermedades infecciosas sobre la base de la diferenciación molecular de cepas. Este esquema permite la caracterización genotípica rápida de microorganismos, gracias al desarrollo de una base de datos centralizada (PubMLST) que permite comparar diversas variantes genéticas denominadas tipos de secuencias (ST, por sus siglas en inglés, sequence type,) y delinear rutas potenciales de dispersión 5.
El objetivo del presente estudio fue determinar las variantes genéticas de los aislados patógenos de la V. parahaemolyticus asociadas a casos humanos y alimentos marinos circulantes en el Perú entre 1995-2017, usando la técnica del MLST y detección in silico de genes de virulencia.
MENSAJES CLAVE
Motivación para realizar el estudio: Ante la eventual emergencia mundial de patógenos causantes de infecciones gastrointestinales, reforzar la vigilancia epidemiológica molecular de microorganismos como la V. parahaemolyticus contribuirá en la detección oportuna y control de brotes.
Principales hallazgos: Se detectaron 15 diferentes genotipos de V. parahaemolyticus, de los cuales, tres de ellos ya han causado importantes brotes en Perú, mientras que los otros 12 tienen potencial para causar epidemias a futuro debido a su carácter virulento.
Implicancias: Se actualiza la información sobre los genotipos circulantes en el Perú hasta el 2017, su prevalencia y distribución a través del tiempo.
EL ESTUDIO
Se incluyeron 16 cepas remitidas como V. parahaemolyticus pertenecientes a la colección del Laboratorio de Referencia Nacional de Enteropatógenos del Instituto Nacional de Salud (INS) (Bioproject: PRJNA556706) y 86 genomas peruanos disponibles en la base de datos del NCBI ( http://www.ncbi.nlm.nih.gov) para el análisis MLST (Tabla 1).
Tabla 1 Tabla de datos de Vibrio parahaemolyticus peruanos utilizados en este estudio.
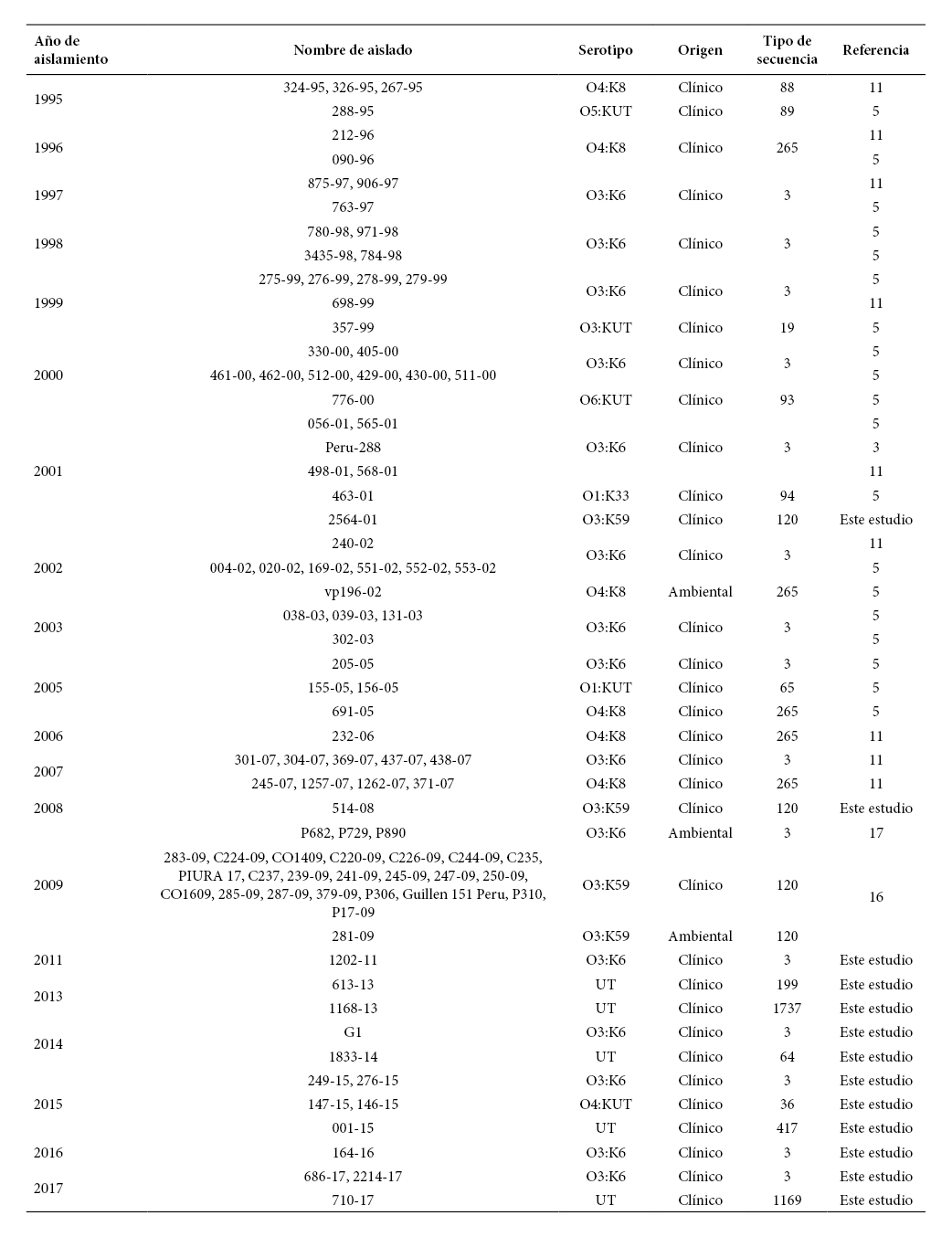
UT: No se pudo tipificar.
Las cepas fueron recuperadas en agua de peptona alcalina (Merck, Alemania) a 37 °C durante 68 horas. Posteriormente, fueron sembradas por estría en placas de agar tiosulfato citrato bilis esculina (Merck, Alemania), y se incubaron a 37 °C durante 18 a 24 horas. El género Vibrio fue confirmado utilizando pruebas bioquímicas convencionales, y la especie V. parahaemolyticus mediante PCR por la presencia del gen toxR descrito por Kim et al. 6
La extracción del ADN se realizó utilizando el kit DNeasy Blood &Tissue (Qiagen, Alemania). La concentración y pureza del ADN se evaluaron mediante espectrofotometría (Denovix, EE. UU). La elaboración de bibliotecas de secuenciamiento se realizó con el kit Nextera XT (Illumina, EE. UU), y el secuenciamiento genómico, con el secuenciador de alto rendimiento MiSeq (Illumina, EE. UU) 7. La calidad de las secuencias obtenidas fue evaluada mediante el programa FastQC v0.11.5. Las secuencias se ensamblaron de novo empleando el pipeline A5-miseq 8. La identificación del género y la detección de contigs contaminados se realizó con el programa Kraken 9.
Los perfiles alélicos y los ST de los genomas obtenidos se asignaron de acuerdo a la información de la base de datos del MLST para V. parahaemolyticus ( ), utilizando el programa MLST v2.10, sobre la base del esquema de siete locus descrito para la V. parahaemolyticus 5. La asignación de complejos clonales se realizó utilizando el programa BioNumerics v7.5 (Applied Maths). La inclusión en un complejo clonal (CC) se restringió a los ST que compartían al menos 6 de los 7 alelos, mientras que los singletons se definieron como ST que difieren en dos o más alelos de los otros STs. Con el mismo programa se generó un minimum spanning tree (MST) que muestra los ST y los CC incluidos en este trabajo. Adicionalmente, se construyó una gráfica de barras apiladas de los aislados estudiados empleando Infostat, para visualizar genotipos por año de aislamiento y por número de muestras.
Se utilizó la herramienta BLASTn para buscar factores de virulencia de la V. parahaemolyticus: la isla de patogenicidad de tipo 7 (VpaI-7) que contiene internamente la variante más común de la TDH obtenida del cromosoma 2 del genoma RIMD 2210633 (número de acceso: NC_004605.1) y la TRH obtenida del aislado AQ4299 (número de acceso: LC271586.1), identificando como homólogos aquellos que presenten <90% de identidad y una cobertura <60% de alineamiento a la referencia. El código empleado para la anotación se encuentra disponible en . Los resultados obtenidos fueron ordenados en formato de tabla en la que se indica la presencia o la ausencia de genes. Todas las secuencias obtenidas durante el estudio han sido depositadas en el GenBank (número de acceso de Bioproject: PRJNA556706).
EL HALLAZGO
Las 16 cepas fueron confirmadas como V. parahaemolyticus por la presencia del gen ToxR. En cuanto a la información genómica, se obtuvo, en promedio, un tamaño genómico de 5,18 pb, compuestas por 80 contigs y un porcentaje de GC de 45,2%.
En total, se analizaron 102 genomas de cepas peruanas de origen clínico (97) y ambiental (5), aisladas en el periodo 1995-2017 (Tabla 1). Los 102 genomas fueron clasificados en 15 ST diferentes, que fueron agrupados por año de aislamiento y origen del aislado (Figura 1). Se observa que no se obtuvieron aislados en los años 2004, 2010 y 2012.
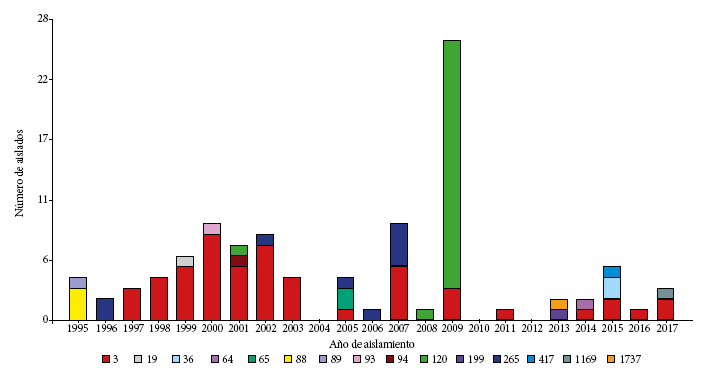
Figura 1 Distribución de Vibrio parahaemolyticus por año de aislamiento, elaborado con el programa InfoStat. La leyenda inferior indica el color según el tipo de secuencia.
La estructura poblacional de las cepas de la V. parahaemolyticus peruanos (n=102) analizadas por MLST se puede visualizar mediante un MST (Figura 2). Todas las cepas pertenecientes al complejo pandémico O3:K6 se agruparon en el ST3 (n=53), correspondiente al 52% de las cepas analizadas. Además, se identificó el complejo clonal 345 (CC345) compuesto por los ST88 (n=3) y ST265 (n=9), ambas del serotipo O4:K8 que difieren en un solo locus, con una frecuencia de aislamiento de 11,8%. Las cepas restantes se incluyeron en 12 ST no relacionados, resaltando al ST120 23,5% (n=24) y ST36 1,9% (n=2) por estar relacionados con brotes o epidemias a nivel local o global (Anexo 1).
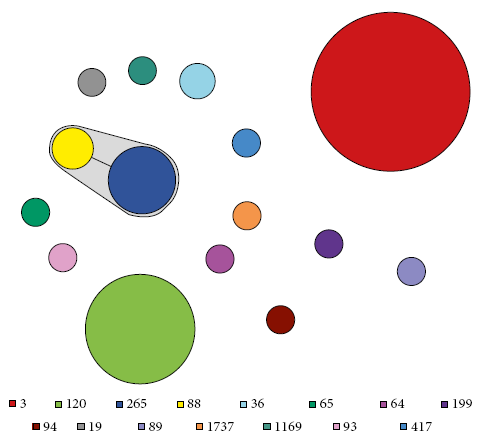
Figura 2 Minimum spanning tree (MST) de 102 perfiles alélicos de MLST de V. parahaemolyticus incluidos en este estudio diseñado con el programa BioNumerics v7.5. La leyenda indica cada tipo de secuencia, diferenciado por colores. Cada círculo representa un genotipo MLST y el tamaño es proporcional al número de cepas que incluye cada uno. Las ramas que conectan los círculos indican que pertenecen al mismo complejo clonal.
De los 102 genomas analizados, 89 aislados poseían la isla de patogenicidad VpaI-7 (87,3%). Las copias de los genes que codifican la TDH se encontraron en 96 aislados (94,1%), siendo los ST más frecuentes: ST3, ST36, ST88, ST120 y ST265, mientras que los genes de la TRH se encontraron únicamente en los ST36, ST64, ST65 y ST417, dando un total de 6 aislados (5,9%). Los resultados agrupados por ST se pueden visualizar en la Tabla 2, mientras que los resultados por aislado de los genes que componen la VpaI-7 se pueden ver en el Anexo 2.
Tabla 2 Detección in silico de factores de virulencia de V. parahaemolyticus por tipo de secuencia
| Tipo de secuencia | n | VpaI-7 | tdhA | tdhS | trh |
|---|---|---|---|---|---|
| ST3 | 53 | 53 | 53 | 53 | 0 |
| ST19 | 1 | 0 | 0 | 0 | 0 |
| ST36 | 2 | 0 | 2 | 2 | 2 |
| ST64 | 1 | 0 | 1 | 1 | 1 |
| ST65 | 2 | 0 | 0 | 0 | 2 |
| ST88 | 3 | 3 | 2 | 2 | 0 |
| ST89 | 1 | 0 | 1 | 1 | 0 |
| ST93 | 1 | 0 | 1 | 1 | 0 |
| ST94 | 1 | 0 | 0 | 0 | 0 |
| ST120 | 24 | 24 | 23 | 23 | 0 |
| ST199 | 1 | 0 | 1 | 1 | 0 |
| ST265 | 9 | 9 | 9 | 9 | 0 |
| ST417 | 1 | 0 | 1 | 1 | 1 |
| ST1169 | 1 | 0 | 1 | 1 | 0 |
| ST1737 | 1 | 0 | 1 | 1 | 0 |
| Total | 102 | 89 | 96 | 96 | 6 |
n: número de aislados, VpaI-7: isla de patogenicidad 7, tdhA: gen de la hemolisina directa termoestable A, tdhS: gen de la hemolisina directa termoestable S, trh: gen de la hemolisina relacionada a la TDH.
DISCUSIÓN
V. parahaemolyticus es un patógeno transmitido por alimentos de alto consumo, y, a pesar de ello, se tiene poca información sobre las variantes patogénicas y la prevalencia temporal de sus genotipos en el Perú. Este reporte subestimado de infecciones por V. parahaemolyticus en el ámbito hospitalario se debe a las deficiencias en el monitoreo y la investigación de enfermedades transmitidas por alimentos 10.
Analizando la prevalencia temporal de genotipos detectados, los aislados con mayor antigüedad corresponden a los años 1995-1996, en especial al serotipo O4:K8, pero de genotipos distintos: ST88 y ST265. Estudios previos reportan a este serotipo desde 1980 causando casos esporádicos y pequeños brotes asociado al consumo de alimentos marinos crudos, siendo en 1983 el más prevalente 11. Estudios de vigilancia molecular revelaron a China como el origen de este serotipo, compuesto por diversos genotipos que no tienen relación clonal con el serotipo O3:K6, sugiriéndolo como un importante complejo clonal, CC345. Además, análisis de genómica comparativa revelaron que estos aislados presentaban las regiones del sistema de secreción tipo 3 (T3SS) y la VpaI-7 12, lo cual es acorde a lo encontrado aquí.
Por otro lado, la presencia del ST3 resalta en la distribución por año y en el MST, al ser el genotipo con mayor número de aislados secuenciados, abarcando cepas aisladas entre 19972017. Todas ellas presentaron la VpaI-7 completa. El primer brote peruano del ST3 se produjo en 1997; sin embargo, se conoce que emergió en la India en 1996 expandiéndose hasta alcanzar el continente americano 13. Este genotipo pertenece al complejo clonal pandémico (CC3) de amplia distribución mundial, siendo el clon dominante hasta hoy 5, además de ser el genotipo más explorado por poseer la mayoría de VpaI reportadas para V. parahaemolyticus, siendo la VpaI-7 la más importante, asociada a citotoxicidad y enterotoxicidad por la presencia de la TDH y el T3SS 14.
El ST120 es el segundo grupo con mayor número de aislados en este estudio, obtenidos mayormente durante el 2009 debido a un brote del serotipo O3:K59 en el norte del país 15, el cual luego de la aplicación de vigilancia molecular por MLST fue definido como ST120, cuyo origen es China, representando la tercera introducción de poblaciones patógenas de V. parahaemolyticus 16. En los resultados, llama la atención la presencia de este genotipo durante 2001, muchos años antes de su primer reporte, lo cual permitiría replantear la introducción de este genotipo al Perú. Estudios de genómica comparativa serían necesarios para encontrar diferencias entre estos aislados y los que causaron el brote del 2009, los cuales comparten la presencia de la VpaI-7.
Adicionalmente, el ST36, un grupo pequeño de importancia epidemiológica dentro de los aislados peruanos analizados, tiene su origen en la región noroeste del pacífico de Norteamérica causando brotes en los Estados Unidos y Canadá 1, el cual se ha expandido desde el 2012 hasta alcanzar rápidamente otras áreas geográficas como el noroeste de España 17. En base a nuestros resultados, las primeras cepas pertenecientes a este ST aparecen en Perú en el 2011, siendo parte de la expansión de este clon en el Pacífico 18, y aunque no se ha reportado ningún brote de este genotipo, representa un riesgo epidemiológico latente debido a su potencial patogénico por la presencia de la TDH y la TRH. En este aspecto, la vigilancia molecular por MLST puede ser aplicada para el rastreo oportuno de estos aislados y frenar brotes.
En cuanto a los otros ST, no se encontró información que los señale como causantes de brote o de las epidemias alrededor del mundo. Sin embargo, no se descarta su potencial patogénico, debido a la presencia de genes que codifican la TDH o la TRH, resultados que incluyen genotipos como los ST1169 y ST1737.
El análisis de factores de virulencia detectó la presencia de los genes que codifican la TDH en gran parte de aislados clínicos, particularmente en los genotipos con mayor número de aislados analizados. Se conoce que los genes que codifican la TDH mayormente se ubican dentro de la VpaI-7, por lo que la presencia de esta región tendrá un impacto en el incremento de la virulencia de V. parahaemolyticus 19; sin embargo, la deleción genética de las copias del gen tdh o de la VpaI-7 completa no es determinante para la ausencia de virulencia 14, lo que explica la detección de cepas clínicas con ausencia de TDH. Por otro lado, se detectaron genomas con TDH pero sin VpaI-7, lo cual ya fue descrito como variantes TDH no asociadas a VpaI-7 20. Finalmente, el gen que codifica la TRH, el cual causa un efecto similar a la TDH, se encontró en un grupo muy reducido de aislados.
En conclusión, el ST3 predomina tanto temporal como numéricamente en el Perú, por lo que hasta el día de hoy es un genotipo de riesgo en salud pública asociado al consumo de alimentos marinos crudos o semicrudos, destacando su potencial patógeno debido a la presencia de la VpaI-7 portadora de hemolisinas. Esto, sumado a la data epidemiológica subestimada, así como la circulación de otras variantes patógenas de esta bacteria en el país, sugiere incrementar los esfuerzos en la vigilancia molecular, la cual está resultando ser una herramienta poderosa para detectar y controlar brotes e infecciones.

