










Services on Demand
Journal

Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Peruana de Medicina Experimental y Salud Publica
Print version ISSN 1726-4634On-line version ISSN 1726-4642
Rev. perú. med. exp. salud publica vol.38 no.2 Lima Apr./Jun. 2021 Epub Mar 30, 2021
http://dx.doi.org/10.17843/rpmesp.2021.382.6417
Originales breves
Using SARS-CoV-2 genomic information to estimate the effective reproductive number (Rt) in Peru during March - April 2020
, Biologist with specialization in Genetics and Cell Biology, Doctor in Natural Sciences http://orcid.org/0000-0001-9947-3868
http://orcid.org/0000-0001-9947-3868
, Bachelor of Sciencehttp://orcid.org/0000-0003-2113-0253
, Biologist, Microbiologist, Parasitologisthttp://orcid.org/0000-0002-8147-8318
, Degree in Biology, Doctor of Philosophy in Biology and Biomedical Scienceshttp://orcid.org/0000-0002-1669-2553
1 Departamento de Ciencias Biológicas y Fisiológicas. Facultad de Ciencias y Filosofía, Universidad Peruana Cayetano Heredia, Lima, Perú.
2 Facultad de Ciencias y Filosofía, Universidad Peruana Cayetano Heredia, Lima, Perú.
3 Unidad de Investigación en Enfermedades Emergentes y Cambio Climático, Facultad de Salud Pública y Administración, Universidad Peruana Cayetano Heredia, Lima, Perú.
4 Departamento de Ciencias Celulares y Moleculares, Facultad de Ciencias y Filosofía, Universidad Peruana Cayetano Heredia, Lima, Perú.
The understanding of COVID-19, caused by the SARS-CoV-2, is essential to improve evidence-based public health policies. The effective reproductive number (Rt) in Peru was estimated using information from 113 complete genomes sequenced by the Instituto Nacional de Salud del Perú (INS), available in the GISAID public database. The Rt trend during March and April of 2020 was found to be similar to results from other epidemiological reports. The Rt decreased during the first two weeks of March. Its lowest value was reported during the week after the quarantine began. The Rt increased moderately after the second week of April. The implication of early decisions taken to mitigate the transmission are discussed. Genomic surveillance will be necessary to understand the transmission and evolution of SARS-CoV-2 in Peru, and will complement the epidemiological information.
Keywords: Bayesian Analysis; Nucleotide Databases; COVID-19; Molecular Epidemiology; Phylogenomics; Genomics; Epidemiological Models; Peru; SARS-CoV-2; Surveillance
INTRODUCTION
The COVID19 pandemic is caused by SARS- CoV-2 which was first reported in December 2019 in Wuhan, China. As of February 2021, it has become a global public health problem, affecting more than 111 million people, and resulting in more than 2.4 million deaths 1. Peru reported its first case on March 6, 2020. Ten days later, the government decreed a state of national emergency that included quarantine measures and a curfew 2. However, transmission was widespread within the country. In addition, the number of positive cases by molecular tests increased to more than 364,000 and by rapid tests to more than 840,000 3.
Epidemiological tools are of great importance during a pandemic, especially during the decision-making process, because they can provide a picture of the initial and current transmission dynamics. The use of these tools is supported by the early isolation of positive cases, contact tracing, and selection of appropriate community and individual mitigation measures 4. One of the most important estimates during the early stages of the pandemic is the basic reproductive number (R0) 5 , 6, which represents the number of secondary cases expected from a primary case in a population in which all individuals are susceptible to the disease; it measures the potential speed of transmission during the initial phase of the pandemic. R0 was estimated at 2.3 (95% confidence interval [95% CI]: 2.0-2.5) for Lima 2 ) and between 2.36 and 5.24 for Peru 6 ) during March 2020.
Another important parameter is the effective reproductive number (Rt), which measures the expected number of new infections caused by an infectious individual during the course of the pandemic, when there are already non-susceptible individuals in the population because they were already infected or are immune to the disease 7. If Rt>1, the cases will increase exponentially; if Rt<1, the number of infected individuals will decrease 8. The Rt for Peru was estimated at 2.36 (95% CI: 2.11- 2.63) during March 2020 7.
The availability of free data in public repositories encourages open science and rapid public health decision-making 9. For example, the first SARS-CoV-2 genome, published in January 2020, enabled the early development of RT-PCR molecular tests 10, which are used worldwide to confirm the presence of the virus. Genomic information is also used in phylodynamic analyses to obtain estimates of the transmission of emerging diseases, and in real-time molecular evolution processes, thus collaborating with epidemiological surveillance 11.
Rt was calculated with models that are similar to the epidemiological SIR (susceptible, infected, recovered), using phylogenetic information to obtain transmission and recovery rates 12. The first genome of a Peruvian case was sequenced by the Instituto Nacional de Salud (INS) in March 2020 13. As of February 2021, there were 113 Peruvian case genomes from March and April 2020 deposited in the public database Global Initiative on Sharing All Influenza Data (GISAID).
The aim of our study was to use the genomic information available from Peru to estimate the Rt during the first two months of the epidemic and to determine the dynamics of initial transmission by integrating molecular analyses with epidemiological estimates.
KEY MESSAGES
Motivation for the study: The use of coronavirus genomic information can contribute to its epidemiological surveillance in Peru and provide independent data on how the transmission and evolution of the disease occurs.
Main findings: Using genomic information, we found that disease transmission decreased after the beginning of the quarantine. However, it increased after a few weeks, probably due to the difficulty of maintaining a strict quarantine in our society.
Implications: Genomic surveillance of the virus will be useful to monitor the speed of transmission of the new coronavirus at the national level and to assess the effects of new local variants.
THE STUDY
This was a descriptive study. The information was obtained from the GISAID database in February 2021 and corresponds to March and April 2020. The unit of analysis was each of the genomes available for that period. We used 113 genomes that met the following criteria: complete genome (>29,000 base pairs) and a percentage of ambiguous nucleotides of less than 1%. Accession numbers can be found in Supplementary Material 1. Of the samples analyzed, 59 were from Lima and 54 from other regions: 8 from Áncash, 7 from Arequipa, 2 from Cajamarca, 2 from Callao, 2 from Cusco, 1 from Huánuco, 5 from Ica, 7 from Junín, 3 from La Libertad, 12 from Lambayeque and 5 from Loreto. The unit of time for the analysis was a calendar day. Sequences were aligned with the MAFFT program 14 and the SARS-CoV-2 reference genome (GenBank identification code: NC_045512.2). The multiple sequence alignment was manually edited in the Geneious v.2020.2.3 program to remove the flanks corresponding to the untranslated regions (UTRs). The final alignment size resulted in 29,409 sites. This alignment can be found in Supplementary Material 2.
Estimation of Rt from genetic data is possible using the Birth-Death Skyline (BDSKY) model 15. The model is based on the birth-death process, in which individuals generate new disease transmission events at a rate λ (birth) and become noninfectious at a rate δ (death), due to host recovery, behavioral changes, or successful treatment. The effective reproductive number estimated from the genomes is calculated by dividing both rates.
The parameters needed to model these rates were estimated from phylogenies generated by Bayesian analysis in the BEAST2 program 16. This program uses Markov chain Monte Carlo (MCMC) and estimates the parameters of the BDSKY model at each generation of the MCMC. Ten MCMCs were run, with 50 million generations each. Each chain was sampled every 50,000 generations, the first 25% of the results from each chain were discarded (burnin). The results of the 10 MCMCs were combined into a single file (Supplementary Material 3).
From an initial (a priori) distribution, and a significant number of generations, the values tend to appear in a stationary (posterior) distribution. When this happens, the parameters are said to have achieved “convergence”. To accept the convergence of MCMC, 200 or more is the needed estimated sample size (ESS) in the posterior distribution.
A nucleotide substitution rate of 22,905 substitutions per year was used to calibrate the phylogenies. This rate was obtained from the NextStrain genomic surveillance portal 17. The result of the division between the rate and the size of the reference genome (29,903 nucleotides) was 0.00077 substitutions/site/year. Similar mutation rates have been used in other studies on introductions and early SARS-CoV-2 transmission 18 , 19.
The a priori parameters are shown in Table 1. We carried out the following: 1) Calculation of the rate of becoming non-infectious (becomeUninfectiousRate), expressed in units per year, and resulting from dividing the days of the year by the average time of days during which an individual is infectious (365/15 ~25 units). We chose 25 as the initial mean of a normal distribution. 2) The common ancestor of the Peruvian samples was estimated between late December 2019 and January 2020; therefore, we chose a normal distribution with mean 0.3 (~110 days ago) as the initial distribution. 3) The Rt was estimated using a lognormal distribution with mean 1.65 and standard deviation 1 as the parameter, which implies 0.1<Rt<7.1. The Rt was calculated for each week, approximately.
Table 1 A priori parameters of the birth-death skyline model in BEAST2.
| Parameter | Mean | Standard deviation | Distribution |
|---|---|---|---|
| BecomeUninfectiousRate | 25 | 5 | Normal |
| Common ancestor of the Peruvian samples | 0.3 a | 0.05 | Normal |
| Effective reproductive number | 1.65 | 1.00 | Lognormal |
a December 2019 to January 2020
The information used in this study comes from the public database GISAID. None of the data analyzed had any personal identification. This research is aligned with the need for information to address a health emergency.
FINDINGS
The stationary distribution of MCMCs, with convergence (ESS > 200), was achieved by estimating the Rt (Table 2, Supplementary Material 3). We calculated the rate of becoming non-infectious at 39.7, HPD 95%: 31.76-47.62 (HPD: highest probability density interval). This value corresponds to an estimated 365/39.7 ~9 days, HPD 95%: 7.66 to 11.5 days, in which the individual would remain infectious. The Rt was greater than 1 day before the first report of SARS-CoV-2 in Peru (Figure 1, Supplementary Material 4). Rt decreased during the second week of March and reached an Rt<1 value a few days before the start of quarantine. The lowest Rt values were found between March 19 and 25 (Supplementary Material 4), after the quarantine was decreed. Rt fluctuated close to 1 during the following weeks until rising again in the second half of April 2020.
Table 2 Parameters estimated using the Birth-Death Skyline model in BEAST2.
| Parameter | Mean | Interval at HPD 95% | ESS |
|---|---|---|---|
| Posterior probability | -41,340.88 | -41,376.93 - -41,307.96 | 5,261 |
| BecomeUninfectiousRate | 39.7 | 31.76 - 47.62 | 7,023 |
| Origin a | 0.3 | 0.29 - 0.30 | 7,207 |
a Date of occurrence of the common ancestor of the samples.
ESS: estimated sample size. HPD: highest probability density interval
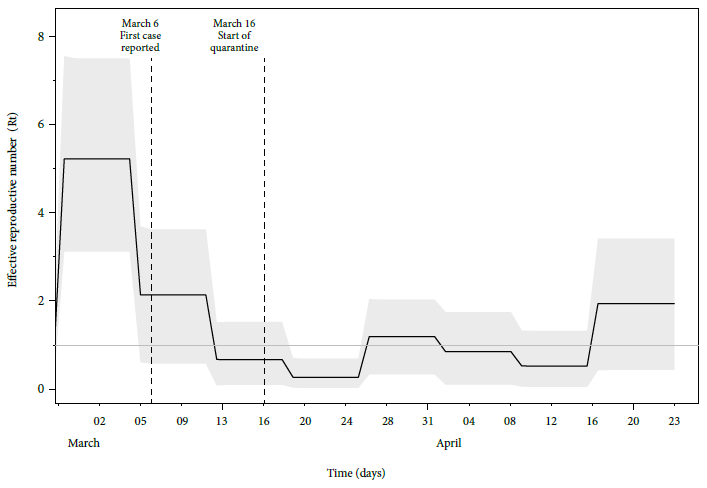
Figure 1 Estimated values of Rt by the Birth-Death Skyline model. Uncertainty is represented by a distribution with higher probability density intervals at 95% (HPD 95%) (shaded area). The Rt value decreases during the first two weeks of March and increases moderately during the last week of April 2020.
DISCUSSION
During the first ten days of pandemic in Peru, the Rt reported was 2.36-5.24 7. Another report calculated an Rt of 4.4 for March 2020 and a reduction to 3.2 during the first two weeks of quarantine 4. Our research shows concordance with these estimates and with non-peer-reviewed reports 8. The higher initial values could be due to initial uncertainty in the calculation of Rt during the first few days 8.
Control of the epidemic can be achieved by reaching a value of Rt<1. According to our results, the quarantine allowed Peru to maintain this value for at least one week after the quarantine was decreed. However, the decrease in Rt before the beginning of the quarantine could be explained by the measures taken by the government the previous week, such as suspending classes and flights from Europe and Asia, as well as prohibiting meetings of more than 300 people 2.
Rt<1 was maintained during the first week of quarantine probably due to the strict conditions applied during the first days. The subsequent increase in Rt after that week may have been caused by recurrent crowding in public places such as markets or banks. For example, the failure of the sex-specific mobilization restriction (April 3-10, 2020), which caused crowding on days intended for females.
Although early strict mitigation measures such as confinement were taken, it did not prevent people from moving out of their homes. Confinement has worked in countries with formal labor markets, but in Peru, the suppression of informal economic activities — which reaches more than 72% — is an unsustainable measure in the context of our social dynamics 20, that may have contributed to people leaving their homes and enabled the transmission of the virus.
Regarding the limitations of our study, the number of genomes that were sampled is small compared to the total cases reported in Peru during March and April 2020. However, other studies have used similar methodologies to estimate the Rt, assuming a sampling frequency of 10-3 and 10-5 (1/1,000 to 1/100,000) genomes per reported cases 18 , 19. At the end of April 2020, there were more than 36,000 cases, so the sampling frequency in Peru would be 113/36,000 ~10-3. Even if this official figure was lower than the actual existing cases, the value would still be within the range used in other studies.
On the other hand, geographic representation should also be improved to determine the value of the Rt at the regional level with a greater detail. This is another limitation of our study and is related to the public data available in the GISAID database. Therefore, we suggest increasing the geographic diversity of the reported samples.
In conclusion, by using genomic information, it is evident that the Rt decreased considerably during the first half of March, reached its lowest value the week after the beginning of quarantine, and then increased moderately since the second half of April.
It is clear that more sequencing initiatives are needed to strengthen genomic surveillance in our country. The power of this analysis facilitates follow-up of new mutations, foreign variants, and local variants and provides data on the genetic diversity of the virus, its molecular evolution and its phylogeographic transmission patterns in our country. In this study, the genomic information obtained was analyzed with an epidemiological approach to estimate the speed of virus transmission during the early stage of the pandemic.
Since genomic data complement epidemiological studies, their integration is necessary for a future national epidemiological surveillance system that will allow public health decisions to be made based on evidence provided by open and multidisciplinary science. It is also necessary to promote the training of skilled human resources who can understand and integrate epidemiological methods with bioinformatics tools.
REFERENCES
. WHO. Coronavirus Disease (COVID-19) Dashboard [Internet]. Suiza: WHO; 2020 [citado el 2 setiembre de 2020]. Disponible en: https://covid19.who.int. [ Links ]
. Munayco CV, Tariq A, Rothenberg R, Soto-Cabezas GG, Reyes MF, Valle A, et al. Early transmission dynamics of COVID-19 in a southern hemisphere setting: Lima-Peru: February 29(th)-March 30(th), 2020. Infect Dis Model. 2020;5:338-45. doi: 10.1016/j.idm.2020.05.001. [ Links ]
. MINSA. Sala situacional COVID-19 [Internet]. Perú: Ministerio de Salud; 2020 [citado el 02 setiembre 2020]. Disponible en: https://covid19.minsa.gob.pe/sala_situacional.asp. [ Links ]
. Gonzales-Castillo JR, Varona-Castillo L, Dominguez-Morante MG, Ocaña-Gutierrez VR. Pandemia de la COVID-19 y las Políticas de Salud Pública en el Perú: marzo-mayo 2020. Revista de Salud Pública. 2020;22(2):1-9. doi: 10.15446/rsap.v22n2.87373. [ Links ]
. Ridenhour B, Kowalik JM, Shay DK. Unraveling R0: considerations for public health applications. Am J Public Health. 2014;104(2):e32-41. doi: 10.2105/AJPH.2013.301704. [ Links ]
. Torres-Roman JS, Kobiak IC, Valcarcel B, Diaz-Velez C, La Vecchia C. The reproductive number R0 of COVID-19 in Peru: An opportunity for effective changes. Travel Med Infect Dis. 2020:101689. doi: 10.1016/j.tmaid.2020.101689. [ Links ]
. Caicedo-Ochoa Y, Rebellon-Sanchez DE, Penaloza-Rallon M, Cortes-Motta HF, Mendez-Fandino YR. Effective Reproductive Number estimation for initial stage of COVID-19 pandemic in Latin American Countries. Int J Infect Dis. 2020;95:316-8. doi: 10.1016/j.ijid.2020.04.069. [ Links ]
. Rt de COVID-19 por departamento en Perú [Internet]. Lima: AMIGOCLOUD; 2020 [citado el 5 setiembre de 2020]. Disponible en: https://huaynodata.com/. [ Links ]
. Moorthy V, Henao Restrepo AM, Preziosi MP, Swaminathan S. Data sharing for novel coronavirus (COVID-19). Bull World Health Organ. 2020;98(3):150. doi: 10.2471/BLT.20.251561. [ Links ]
. Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DK, et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25(3). doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [ Links ]
. Rife BD, Mavian C, Chen X, Ciccozzi M, Salemi M, Min J, et al. Phylodynamic applications in 21(st) century global infectious disease research. Glob Health Res Policy. 2017;2:13. doi: 10.1186/s41256-017-0034-y. [ Links ]
. Vaughan TG, Leventhal GE, Rasmussen DA, Drummond AJ, Welch D, Stadler T. Estimating Epidemic Incidence and Prevalence from Genomic Data. Mol Biol Evol. 2019;36(8):1804-16. doi: 10.1093/molbev/msz106. [ Links ]
. Padilla-Rojas C, Lope-Pari P, Vega-Chozo K, Balbuena-Torres J, Caceres-Rey O, Bailon-Calderon H, et al. Near-Complete Genome Sequence of a 2019 Novel Coronavirus (SARS-CoV-2) Strain Causing a COVID-19 Case in Peru. Microbiol Resour Announc. 2020;9(19). doi: 10.1128/MRA.00303-20. [ Links ]
. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019;20(4):1160-6. doi: 10.1093/bib/bbx108. [ Links ]
. Stadler T, Kuhnert D, Bonhoeffer S, Drummond AJ. Birth-death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc Natl Acad Sci U S A. 2013;110(1):228-33. doi: 10.1073/pnas.1207965110. [ Links ]
. Bouckaert R, Vaughan TG, Barido-Sottani J, Duchene S, Fourment M, Gavryushkina A, et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2019;15(4):e1006650. doi: 10.1371/journal.pcbi.1006650. [ Links ]
. Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34(23):4121-3. doi: 10.1093/bioinformatics/bty407. [ Links ]
. Lai A, Bergna A, Acciarri C, Galli M, Zehender G. Early phylogenetic estimate of the effective reproduction number of SARS-CoV-2. J Med Virol. 2020;92(6):675-9. doi: 10.1002/jmv.25723. [ Links ]
. Lai A, Bergna A, Caucci S, Clementi N, Vicenti I, Dragoni F, et al. Molecular tracing of SARS-CoV-2 in Italy in the first three months of the epidemic. Viruses. 2020;12(8):798. doi: 10.3390/v12080798. [ Links ]
. Organización Mundial del Trabajo. ¿Respuesta rápida a la COVID-19 en un contexto de alta informalidad? El caso del Perú [Internet]. Ginebra: Organización Mundial del Trabajo; 2020 [citado el 13 de setiembre de 2020]. http://www.ilo.org/wcmsp5/groups/public/ed_emp/documents/publication/wcms_747776.pdf. [ Links ]
Funding: This work was funded by Fondo Nacional de Desarrollo Científico y Tecnológico y de Innovación Tecnológica (Fondecyt-Perú) under “Proyecto de Mejoramiento y Ampliación de los Servicios del Sistema Nacional de Ciencia, Tecnología e Innovación Tecnológica” (Contract No. 34-2019-FONDECYT- BM-INC. INV.), and by CONCYTEC-FONDECYT as part of the contest “Proyectos Especiales: Respuesta al COVID-19 2020-01” (Contract No. 046-2020-FONDECYT).
Received: September 14, 2020; Accepted: March 03, 2021
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
