











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Un nuevo agente infeccioso fue detectado a finales de diciembre de 2019 en la ciudad de Wuhan, capital de la provincia de Hubei en China; fue designado como coronavirus 2 del síndrome respiratorio agudo severo (SARS-CoV-2) responsable de la COVID-19, la cual fue declarada por la Organización Mundial de la Salud (OMS) como pandemia global el 11 de marzo de 2020 1 - 4. En el mundo, según datos epidemiológicos de la OMS, hasta el 16 de noviembre de 2021 existen 250 millones de casos acumulados de COVID-19, y más de cinco millones de muertes, con un incremento importante de nuevos casos 5.
Actualmente no existe una terapia antiviral eficaz para la COVID-19, por lo que es necesario el desarrollo y la producción de vacunas con propiedades inmunogénicas, que permitan la reducción de la transmisión del SARS-CoV-2 y/o las formas graves de la enfermedad 6 , 7. Sin embargo, todas las vacunas contra la COVID-19 deben cumplir con criterios de rigurosidad para su desarrollo durante la fase preclínica y clínica, deben ceñirse al cumplimiento de los estándares de eficacia y seguridad establecidos por la OMS y entidades regulatorias de cada país ( 8.
La FDA (Food and Drug Administration) como entidad regulatoria, ha establecido criterios de clasificación de las reacciones no deseadas, de acuerdo con la intensidad de los eventos específicos en: leve (grado 1); moderado (grado 2); severo (grado 3), y potencialmente mortal (grado 4) esto permite la categorización de los eventos adversos (EA) observados 9.
La identificación de EA serios, como el síndrome de trombocitopenia y trombosis inmunitaria inducida por la vacuna (STTIV), produjo que algunos países limitaran la administración de las vacunas AZD1222 y Ad26.COV2.S, a pesar de la incidencia muy baja de los casos reportados, es indispensable una mayor divulgación de estos acontecimientos a la población 10.
Esta revisión aborda los aspectos más relevantes acerca la eficacia y seguridad de las vacunas en ensayos clínicos de fase III contra la COVID-19 en adultos y niños, haciendo énfasis en las características reactogénicas de cada vacuna. Pero, además, incluye datos importantes de los estudios de vigilancia poscomercialización.
ESTRATEGIA DE BÚSQUEDA
Se realizó una búsqueda de los artículos de investigación relacionados con las vacunas contra el SARS-CoV-2, en la base datos de PubMed/Medline, cuyos descriptores utilizados fueron: COVID-19, SARS-CoV-2, vacuna, seguridad, inmunogenicidad, eficacia y vigilancia; limitando la búsqueda a artículos en inglés.
Se incluyeron estudios de ensayos preclínicos, clínicos y vigilancia poscomercialización; aunque se agregó algunas revisiones descriptivas relacionadas al tema publicados hasta el 9 de noviembre de 2021. La búsqueda arrojó un total de 3155 artículos relacionados con vacunas contra el SARS-CoV-2, de ellos, se seleccionaron aquellos artículos de mayor relevancia, considerando el diseño del estudio, tamaño muestral, análisis estadístico y aleatorización de la muestra, por lo que se incluyeron 58 publicaciones (Figura 1).
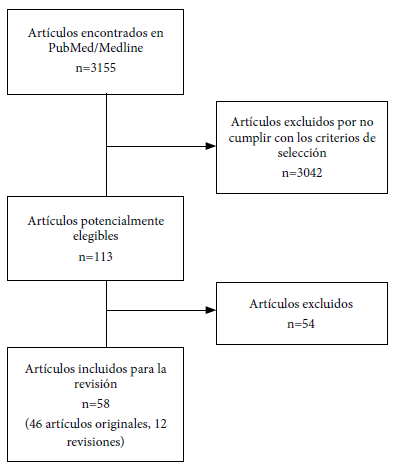
También se revisó la página web de la OMS (https://www.who.int/es/emergencies/diseases/novel-coronavirus-2019/covid-19-vaccines) y la FDA (https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/covid-19-vaccines) donde se incluyeron términos de búsqueda como «vacunas», «COVID-19» y «SARS-CoV-2», encontrando datos epidemiológicos, etapas de desarrollo de las vacunas y normas de regulación, que incluyó cuatro documentos.
VACUNAS EN ENSAYO CLÍNICO DE FASE III Y APROBADAS CONTRA LA COVID-19
Las vacunas contra la COVID-19 representan una alternativa eficaz para la prevención de la infección por el SARS-CoV-2; hasta el 16 de noviembre de 2021 la OMS registró 132 vacunas candidatas en fase de desarrollo clínico y 194 en evaluación preclínica 11. Además, esta organización informa que más de veinte candidatos vacunales se encuentran en ensayos clínicos de fase III (Tabla 1) y algunos fueron aprobados de manera condicional para su comercialización y aplicación en humanos 11 , 12.
Tabla 1 Vacunas en ensayos clínicos de fase III y aprobadas contra la COVID-19.
| Plataforma de vacunas | Desarrollador | Tipo de vacuna candidata | Fase de ensayo clínico/entidad de aprobación | Dosis/ intervalos/vía | Eficacia | Grado de eventos adversos locales y sistémicos |
|---|---|---|---|---|---|---|
| ARNm | BioNTech y Pfizer | ARNm codificante de glicoproteína S encapsulado con nanopartículas lipídicas | Fase II/III OMS EMA FDA | Dos dosis Día 0 y día 21 IM | 91,3% (mayores de 12 años) 90,7% (niños de 5 a 11 años) | Grado 1, 2 y en menor frecuencia 3 |
| ARNm | Moderna/Instituto Nacional de Alergias y Enfermedades Infecciosas (NIAID), de los Estados Unidos | ARNm codificante de glicoproteína S encapsulado con nanopartículas lipídicas | Fase III OMS EMA FDA | Dos dosis Día 0 y día 28 IM | 93,2% (mayores de 18 años) 93,3% (individuos de 12 a 17 años) | Grado 1 y 2 (local) Grado 1,2 y 3 en algunos casos (sistémico) |
| ADN | Inovio Pharmaceuticals | ADN codificante de glicoproteína S contenida en el plásmido pGX9501 con electroporación | Fase II/III | Dos dosis Día 0 y día 28 ID, seguida de EP utilizando el dispositivo CELLECTRA® 2000 | No hay datos | Grado 1 (evaluada en la fase I) |
| Vector viral no replicante | Instituto de Investigación Gamaleya y el Ministerio de Salud de la Federación de Rusia | Adenovirus recombinante tipo 26 y 5 (rAd26 y rAd5) portadores del gen codificante de glicoproteína S | Fase III Rusia, Argentina, Bolivia, Venezuela Paraguay, México, Nicaragua y otros 19 países | Dos dosis Día 0 y día 21 IM | 91,6% (adultos de 18 a 60 años) 91,8% (adultos >60 años) | Grado 1 y 2 |
| Vector viral no replicante | AstraZeneca y Universidad de Oxford | Adenovirus de chimpancé no replicante portador del gen que codifica la glicoproteína S | Fase II/III y fase III OMS EMA FDA | Dos dosis Día 0 y día 28 IM | 70,4% (eficacia general) 62,1% (dosis estándar) 90,0% (dosis baja y estándar) | Grado 1, 2 y 3 |
| Vector viral no replicante | Farmacéutica Janssen y Johnson & Johnson | Adenovirus recombinante tipo 26 (rAd26) | Fase III | Una dosis en día 0 Dos dosis Día 0 y día 56 IM | 66,9% (adultos >18 años) | Grado 1 y 2 (local) Grado 1,2 y 3 (sistémico) |
| Virus inactivado | Sinovac Biotech | Cepa CN02 de SARS-CoV-2 inactivado y adyuvada con hidróxido de aluminio | Fase III NMPA Indonesia, Turquía, Brasil y Chile | Dos dosis Día 0 y día 14 IM | 83,5% (adultos >18 años) | Grado 1, 2 y en menor frecuencia 3 |
| Virus inactivado | Instituto de Productos Biológicos de Beijing y Sinopharm | Cepa HB02 de SARS-CoV-2 inactivado y adyuvada con hidróxido de aluminio | Fase III NMPA Baréin y Emiratos Árabes Unidos | Dos dosis Día 0 y día 21 IM | 86% | Grado 1 y 2 (evaluada en la fase I/II) |
Adaptado del panorama de vacunas contra COVID-19 de la OMS, mayo de 2021. EMA: European Medicines Agency (Agencia Europea de Medicamentos). FDA: Food and Drug Administration (Administración de Medicamentos y Alimentos). NMPA: National Medical Products Administration (Administración Nacional de Productos Médicos). Escala de gravedad: grado 1 (leve), grado 2 (moderado), grado 3 (severo) y grado 4 (potencialmente mortal).
Fuente: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines
Las vacunas contra la COVID-19 están desarrolladas sobre diferentes plataformas que incluyen virus vivos atenuados, virus inactivados, vacunas de subunidades, vacunas de vectores virales no replicantes y replicantes, vacunas de ácidos nucleicos como el ácido desoxirribonucleico (ADN) y ácido ribonucleico (ARN), vacunas de partículas similares a virus (Virus-like particles), entre otros 13. Las vacunas en desarrollo deben ser eficaces (inmunogénicas) y seguras (poco reactogénicas) para facilitar la inmunidad poblacional, siendo este último criterio el más importante para su aprobación 14.
VACUNAS DE ÁCIDO NUCLEICO
Las vacunas de ácido nucleico incluyen las basadas en ARN mensajero (ARNm) y ADN, que introducen el genoma viral dentro la célula eucariota, donde se produce la transcripción y traducción de las proteínas virales que son reconocidas por el sistema inmune 15. Los segmentos del genoma del SARS-CoV-2 empleados para el desarrollo de estas vacunas son los que codifican para la glicoproteína S, el dominio S1 y el dominio de unión al receptor (receptor Binding Domain - RBD) 15 , 16. La secuencia genética completa del SARS-CoV-2 permitió el rápido proceso de desarrollo y producción de estas vacunas, ya que no se requieren cultivar grandes cantidades del virus; sin embargo, este tipo de vacunas no han sido autorizadas para su empleo en humanos antes del inicio de la pandemia, por lo que existe limitada información acerca de su seguridad 17.
VACUNAS BASADAS EN ARNm
Vacuna BNT162b2 (COMIRNATY)
En el ensayo clínico aleatorizado de fase I, la empresa alemana BioNTech y Pfizer evaluaron dos vacunas de ARNm modificado con nucleósidos (1-metil-pseudouridina) y formulada con nanopartículas lipídicas; la vacuna BNT162b1, que codifica el RBD de la glicoproteína S del SARS-CoV-2 y la vacuna BNT162b2 que codifica la glicoproteína S de longitud completa modificado por dos mutaciones de prolina para bloquearlo en la conformación de prefusión 18 , 19.
El ensayo clínico internacional y multicéntrico de fase II/III, que incluyó a 43 448 participantes mayores de 16 años, reportó en el análisis de seguridad con seguimiento de siete días posterior a la aplicación BNT162b2, que los EA locales identificados fueron leves a moderados y que resolvían en uno a dos días, siendo el dolor en el sitio de inyección el más común, seguido de enrojecimiento e hinchazón. Los EA sistémicos observados fueron más frecuentes en adultos menores de 55 años, siendo los más usuales la fatiga, cefalea, escalofríos y mialgia (Tabla 2); aunque se identificaron reacciones adversas graves, estas representaron menos del 2% 20. En este mismo estudio se encontró cuatro eventos adversos serios (EAS) relacionados con la vacuna BNT162b2, después de un seguimiento de dos meses, la reacción más severa fue la arritmia ventricular paroxística 20.
Tabla 2 Comparativa de la proporción de eventos adversos sistémicos de las vacunas BNT162b2 y ARNm-1273 (evaluadas durante siete días después de cada dosis).
| Eventos adversos sistémicos | BNT162b2 (COMIRNATY) (n=8183) | ARNm-1273 (MODERNA) (n=14 677) | ||
|---|---|---|---|---|
| 16 a 55 años | >55 años | 18 a 65 años (n=10 985) | >65 años (n=3692) | |
| Fiebre | 16% | 11% | 1908 (17,4%) | 370 (10,0%) |
| Fatiga | 59% | 51% | 7430 (67,6%) | 2152 (58,3%) |
| Cefalea | 52% | 39% | 6898 (62,8%) | 1704 (46,2%) |
| Escalofríos | 35% | 23% | 5341 (48,6%) | 1141 (30,9%) |
| Mialgia | 37% | 29% | 6769 (61,6%) | 1739 (47,1%) |
| Artralgia | 22% | 19% | 4993 (45,5%) | 1291 (35,5%) |
| Diarrea | 10% | 8% | No | No |
| Vómitos/ náuseas | 2% | 1% | 2348 (21,4%) | 437 (11,8%) |
Fuente: Polack et al. 20 y Baden et al. (29
La información del ensayo de fase III efectuada en Estados Unidos, que incluyó a 2260 participantes de 12 a 15 años, indica que BNT162b2 parece tener un buen perfil de seguridad y reactogenicidad en esta población; ya que la mayoría de los EA no serios locales y sistémicos fueron transitorios, de gravedad leve a moderada. Los EA atribuidos a la vacuna BNT162b2 se observaron en 33 participantes (2,9%), entre los que destacan dolor en sitio de inyección, hinchazón, enrojecimiento, fiebre, fatiga, cefalea, escalofríos, mialgia, artralgia, vómitos y diarrea; aunque no se reportó EAS relacionados con BNT162b2 21.
Los resultados recientes del ensayo de fase I abierta y de la fase II/III aleatorizada, que incorporaron a 50 y 2268 niños de 5 a 11 años de edad, respectivamente; reportan que los EA no serios asociados a BTN162b2, evaluados hasta siete días después de cada dosis, fueron transitorias (1 a 2 días de duración), y la mayoría de gravedad leve a moderada 22. En el ensayo de fase II/III, se observó algunos EA de grado 3 después de cada dosis, en receptores de BNT162b2 (n=1511), como dolor en sitio de inyección (0,6%), fatiga (0,9%), cefalea (0,3%), escalofríos (0,1%) y mialgia (0,1%); además, se informó de cuatro erupciones cutáneas autolimitadas y leves relacionadas a BNT162b2; sin embargo, no se evidenció EAS atribuidos a la vacuna 22.
La eficacia de la vacuna BNT162b2, reportada en los participantes mayores de 12 años sin infección previa por SARS-CoV-2, fue de del 91,3% ([intervalo de confianza, IC] del 95%: 89,0 - 93,2) y en los participantes con y sin evidencia previa de infección por SARS-CoV-2 del 91,1% (IC del 95%: 88,8 - 93,0); por último, la eficacia en niños de 5 a 11 años con o sin evidencia de infección previa por SARS-CoV-2 fue del 90,7% (IC del 95%: 67,4 - 98,3), todas evaluadas a los siete días, posteriores a la segunda dosis 20 - 22.
Vacuna ARNm-1273 (MODERNA)
La vacuna de ARNm-1273 producida por la empresa Moderna de Estados Unidos, está basada en ARNm modificado con nucleósidos y formulada con nanopartículas lipídicas, que codifica la glicoproteína S de longitud completa de SARS-CoV-2 estabilizada en su conformación de prefusión 23. En el ensayo clínico aleatorizado de fase I que incluyó a 45 participantes de 18 a 45 años, los EA locales identificados fueron leves a moderados, y los EA sistémicos fueron más frecuentes y graves con dosis más altas y después de la segunda dosis, siendo la fatiga, los escalofríos, la cefalea, la mialgia y las náuseas observados en más de la mitad de los participantes 24.
El ensayo clínico aleatorizado, doble ciego y controlado con placebo de fase III, realizado en Estados Unidos, que incorporó a 30 420 participantes mayores de 18 años (rango 18 a 95), reportó los EA solicitados en el grupo de ARNm-1273, estos fueron transitorios con una duración media de dos a tres días, generalmente de grado 1 y 2 de gravedad, y de mayor frecuencia en individuos menores de 65 años. Los EA más comunes fueron el dolor en el lugar de la inyección, fatiga y cefalea; aunque se identificaron otros EA sistémicos (Tabla 2), estos fueron más frecuentes después de la segunda dosis de ARNm-1273. Los EA no solicitados relacionados con ARNm-1273 recopilados hasta 28 días después de cada dosis, se observaron en 1242 participantes (8,2%), pero solo en seis personas (<0,1%) se identificaron EAS vinculados a la intervención ( 25.
Los resultados de la etapa ciega del ensayo de fase III revelan que los EA relacionados a ARNm-1273 se observaron en el 13,9% de los participantes (n=15 184), aunque solo en doce individuos (<0,1%) de este grupo se evidenció eventos adversos serios y los EA severos (grado 3) se observaron en 83 participantes (0,5%), por lo que ARNm-1273 mantuvo un buen perfil de seguridad 26. Finalmente, el ensayo de fase II/III que incluyó a 3732 adolescentes de 12 a 17 años reportó que los EA locales y sistémicos recopilados hasta siete días después cada dosis, fueron transitorios con una duración media de cuatro días, la mayoría de grado 1 o 2, siendo los más comunes dolor en lugar de inyección, fatiga, cefalea, escalofríos y mialgia; aunque se evidenció una elevada frecuencia de EA de grado 3 después de la segunda dosis en el grupo de ARNm-1273 (483/2478, [19,5%]). Los EA no solicitados recopilados hasta 28 días después de cada dosis relacionados a ARNm-1273, tuvieron una incidencia moderada (312/2486, [12,6%]), y no se evidenció EAS atribuidos a la vacuna, aunque se requieren estudios adicionales para una interpretación correcta 27.
La eficacia de la vacuna ARNm-1273 en adultos mayores de 18 años fue de 93,2% (IC del 95%: 127,0%-146,8%) para la prevención de COVID-19 y para adolescentes de 12 a 17 años la eficacia de ARNm-1273 fue de 93,3% (IC del 95%: 47,9-99,9), evaluadas después de 14 días posteriores a la segunda dosis 25 , 27.
El estudio de Shimabukuro et al. acerca de la seguridad de las vacunas BNT162b2 y ARNm-en 1273 en mujeres embarazadas, indican que los EA encontrados en esta población son similares al de mujeres no embarazadas; sin embargo, esta investigación está basada en datos pasivos obtenidos de los sistemas de vigilancia de Estados Unidos, por lo que se requieren estudios controlados para confirmar la seguridad de estas vacunas 28.
VACUNAS BASADAS EN ADN
Las vacunas de ADN utilizan un plásmido que lleva incorporado un determinado gen que codifica un antígeno (glicoproteína S, dominio S1 o RBD) del SARS-CoV-2; este segmento génico es añadido al ADN intranuclear y la maquinaria biosintética de la célula se encarga de la transcripción y traducción de esta proteína antigénica, el cual induce una respuesta inmune humoral y celular 29. La vacuna INO-4800 (Inovio Pharmaceuticals) basada en ADN, demostró un adecuado perfil de seguridad e inmunogenicidad en el ensayo de fase I y, actualmente, se encuentran en ensayo clínico de fase II/III 30.
VACUNA DE VECTORES VIRALES NO REPLICANTES
Vacuna Gam-COVID-Vac/rAd26-S+rAd5-S (SPUTNIK V)
La vacuna heteróloga Gam-COVID-Vac, desarrollada por el Instituto de Investigación Gamaleya y el Ministerio de Salud de la Federación de Rusia, está integrada por dos componentes, un vector de adenovirus recombinante tipo 26 (rAd26) y un vector de adenovirus recombinante tipo 5 (rAd5), ambos portadores del gen del SARS-CoV-2 que codifica la glicoproteína S (rAd26-S y rAd5-S), diseñado de esta forma para reducir la inmunidad contra el vector de adenovirus 31.
En los dos ensayos clínicos abiertos no aleatorizado de fase I/II, que incluyó a 38 participantes adultos sanos de 18 a 60 años en cada ensayo, los EA locales identificados fueron leves (grado 1), siendo el dolor en el lugar de la inyección el más común, observado en 44 participantes (58%), seguido de edema (3/76, 3,9%), la hipertermia (3/76, 3,9%) y la hinchazón (1/76, 1,3%) 32. Las reacciones adversas sistémicas reportadas fueron leves a moderadas (grado 1 y 2), caracterizadas por fiebre (38/76, 50%), cefalea (32/76, 42%), astenia (21/76, 28%), mialgia y artralgia (18/76, 24%) y con menor frecuencia, palpitaciones, diarrea, rinorrea, dolor de garganta, pérdida de apetito, malestar general, entre otros 32.
En el ensayo clínico aleatorizado, doble ciego de fase III, que incluyó a 21 977 mayores de 18 años, se valoró la seguridad después de la administración la primera dosis (día 0) de la vacuna (rAd26) y la segunda dosis (día 21) de Sputnik V (rAd5), observando que la mayoría de los EA (locales y sistémicos) fueron de grado 1 y 2, siendo los más frecuentes, dolor en el sitio de inyección, enfermedad similar a la gripe, dolor de cabeza y astenia. Los EAS se observaron en 45 participantes (0,3%) del grupo Gam-COVID-Vac, aunque ninguno estos eventos se relacionaron con la vacuna. La eficacia de Gam-COVID-Vac identificada en el estudio fue del 91,6% (IC del 95%: 85,6-95,2) a partir del día 21 después de la primera dosis, y en los adultos mayores de 60 años se reporta una eficacia de 91,8% (IC del 95%: 67,1-98,3), aunque no existe evidencia de su eficacia contra las variantes de preocupación de SARS-CoV-2 33.
En el estudio observacional efectuado por Babamahmoodi et al. que contó con 13 435 trabajadores de la salud vacunados (rango de edad 19-78 años), reportaron que 3236 (24%) presentaron EA asociados a Gam-COVID-Vac, siendo los más comunes dolor en el lugar de la inyección (56,9%), fatiga (50,9%), dolor corporal (43,9%), cefalea (35,7%), fiebre (32,9%), artralgia (30,3%), escalofríos. (29,8%) y somnolencia (20,3%) con una duración inferior a tres días en la mayoría de los participantes 34. Estos EA fueron más usuales después de la primera dosis y de mayor frecuencia en mujeres, y aunque se identificó anafilaxia en cuatro participantes, es contraproducente relacionarlos con Gam-COVID-Vac por la escasa evidencia y el diseño del estudio 34.
Vacuna AZD1222/ ChAdOx1 nCov-19 (VAXZEVRIA)
La vacuna AZD1222 desarrollada por la empresa farmacéutica AstraZeneca y la Universidad de Oxford, está basada en un vector de adenovirus de chimpancé no replicante, portador del gen de SARS-CoV-2 que codifica glicoproteína S de longitud completa 35.
En el ensayo clínico de fase I/II que incluyó 1077 adultos sanos de 18 a 55 años, los EA locales reportados fueron leves a moderados, siendo el dolor en el lugar de inyección el más común (328/487, 67%), y el enrojecimiento, el calor y la hinchazón fueron menos frecuentes. Las reacciones adversas sistémicas observadas incluyeron, la fatiga (340/487, 70%), mialgia (340/487, 70%), malestar general (340/487, 70%), cefalea (195/487, 41%), escalofríos (272/487, 56%) y sensación de fiebre (250/487, 51%), todas de gravedad leve a moderada, y con reducción significativa de estos eventos después de la administración profiláctica de paracetamol 36.
En los ensayos clínicos de fase II/III realizados en Reino Unido, Brasil y Sudáfrica, la seguridad se estudió en 23 745 participantes mayores de 18 años que recibieron al menos una dosis de la vacuna o placebo, identificándose 175 EAS (84 en el grupo AZD1222 y 91 en el grupo de control) en 168 participantes, aunque solo uno de estos eventos se relacionó con la vacuna 37. La eficacia general reportada fue de 70,4% (IC del 95,8%: 54,8-80,6), pero en el grupo que recibió dos dosis estándar de la vacuna, la eficacia fue del 62,1% (IC del 95%: 41,0-75,7), lo que contrasta con el grupo que recibió una dosis baja como primera dosis de la vacuna, que reportó una eficacia del 90,0% (IC del 95%: 67,4-97,0) 37.
El ensayo clínico de fase III realizado en Estados Unidos, Chile y Perú, que incluyó a 32 451 participantes mayores de 18 años, reportó que 6238 participantes (28,9%) del grupo AZD1222 (n=21 587) presentaron EA relacionados a la intervención, aunque se observó que solo un EAS fue vinculado a AZD1222. En un subgrupo (AZD1222, n=2037) se constató que la mayoría de los EA locales y sistémicos fueron transitorios (1 a 2 días de duración), por lo general leves a moderados, siendo los más frecuentes, el dolor en el sitio de inyección (58,3%), cefalea (50,2%), fatiga (49,7%), mialgia (41.9%), malestar (35,0%), escalofríos (28,2%), náuseas (15,3%) y fiebre (7,0%). Los resultados de este ensayo indican que los EA tromboembólicos observados, como la trombosis venosa profunda, embolia pulmonar y STTIV fueron inusuales (<0,1%) y muy similares en ambos grupos, lo que podría representar estos hallazgos como eventos independientes de AZD1222 38.
Posterior a la aprobación condicional de AZD1222, distintos grupos de investigación en Europa y Canadá reportaron 42 casos STTIV relacionados con esta vacuna, observadas principalmente en adultos menores de 60 años posterior a la administración de la primera dosis y caracterizadas por trombocitopenia y eventos trombóticos como la trombosis del seno venoso cerebral, trombosis de la vena porta, embolia pulmonar, trombosis arteriales, entre otros; aunque es relevante mencionar que estos EAS severos son extremadamente inusuales 39 - 42.
Vacuna Ad26.COV2.S
La vacuna Ad26.COV2.S desarrollada por la empresa farmacéutica Janssen y financiada por Johnson & Johnson, está basada en un vector de adenovirus recombinante de serotipo 26 (Ad26) no replicante, portador del gen de SARS-CoV-2 que codifica la glicoproteína S estabilizada y de longitud completa 43.
En el ensayo clínico de fase I/IIa, que incluyó a 805 adultos asignados en dos cohortes de edad de 18 a 55 años y mayores de 65 años, se reportó que la mayoría de los EA fueron leves o moderados, siendo los más frecuentes el dolor en el lugar de inyección, fatiga, cefalea, mialgia, fiebre y náuseas. Sin embargo, en este ensayo se identificaron reacciones adversas sistémicas de grado 3 en adultos jóvenes que recibieron dosis baja (15/162, 9%) y alta (32/158, 20%) en mayor frecuencia a lo reportado en los adultos mayores (1/161, 1% y 4/161, 2%, respectivamente) 44.
En el análisis general de seguridad del ensayo multicéntrico de fase III en curso, que incluyó 43 783 participantes mayores de 18 años, se observaron EA de grado 3 en 20 participantes del grupo Ad26.COV2.S (n=21 895) atribuidos a la intervención, en este mismo grupo se identificaron siete EAS relacionados a Ad26.COV2.S, incluyendo un caso de trombosis venosa profunda, síndrome de Guillan Barré y pericarditis 45.
En un subgrupo que recibió Ad26.COV2.S. (n=3356) se evidenció que la mayoría de los EA locales y sistémicos solicitados durante siete días después de la vacunación, fueron de grado 1 o 2, siendo los más frecuentes, dolor en el lugar de la inyección (48,6%), cefalea (38,9%), fatiga (38,2%), mialgia (33,2%) y náuseas (4,2%). La eficacia de Ad26.COV2.S a los 14 y 28 días después de su administración para la prevención de COVID-19 moderado a severo fue del 66,9% (IC del 95%: 59,0-73,4) y 66,1% (IC del 95%: 55,0-74,8) respectivamente; con una eficacia para la prevención de COVID-19 crítico del 76,7% (IC del 95%: 54,6-89,1] después de 14 días y 85,4% (IC del 95%:54,2-96,9) posterior a los 28 días de la administración de la vacuna 45.
En el estudio efectuado por Ashrani et al. se evidenció un incremento importante de la incidencia de trombosis del seno venoso cerebral, posterior a la aplicación de Ad26.COV2.S en la población de Olmsted (EE.UU,), lo que sugiere un posible aumento del riesgo de este evento trombótico tras la vacunación con Ad26.COV2.S 46.
La vacuna vectorizada con adenovirus recombinante tipo 5 (Ad5) no replicante, que demostró un buen perfil de seguridad e inmunogenicidad en los ensayos clínico de fase I y II realizados en Wuhan, de la provincia de Hubei en China, también se encuentra en ensayo clínico de fase III 47 , 48. Esta vacuna porta el segmento génico que codifica la glicoproteína S de SARS-CoV-2, y es desarrollada por el Instituto de Biotecnología de Beijing y CanSino Biologics 48.
VACUNAS DE VIRUS INACTIVADO
Los virus inactivados no son infecciosos o patógenos, y se inactivan mediante procedimientos físicos y químicos lo cual reduce la virulencia, pero mantiene estructuras proteicas íntegras que posibilitan una amplia gama de epítopos conformacionales, lo que permite un adecuado estímulo antigénico 49. En el registro de la OMS existen varias vacunas que utilizan esta plataforma que se encuentran en ensayo clínico de fase III.
Vacuna de SARS-CoV-2 completa inactivada (CoronaVac)
Esta vacuna denominada CoronaVac, desarrollada por Sinovac Biotech, está basada en la cepa CN02 de SARS-CoV-2 inactivado y adyuvada con hidróxido de aluminio 50. El ensayo clínico de fase I/II realizado en China, que incluyó a 744 adultos sanos de 18 a 59 años, reportó que la mayoría de los eventos adversos identificados fueron leves y transitorios, aunque se observó un caso grave de urticaria 51.
En otro estudio de fase I/II incluyó a 422 adultos sanos mayores de 60 años, se observó que los EA no serios solicitados fueron leves y moderados, evaluados durante los 7 días posteriores a la vacunación, siendo el dolor el lugar de inyección (39/421, 9%) el evento local más frecuente, y la fiebre (14/421, 3%) el evento adverso sistémico más común 52.
En el ensayo de fase III realizado en Turquía, que incluyó a 10 218 participantes mayores de 18 años, se evaluó la seguridad en el grupo de CoronaVac (n=6646) y placebo (n=3568), observando que la mayoría de los EA identificados fueron leves (grado 1) y transitorios, aunque se observó 25 EA de grado 3 en 22 sujetos (0,3%) del grupo de la vacuna, y solo un EAS clasificado como severo relacionado con CoronaVac, lo que permite sugerir que esta vacuna es segura. La incidencia de EA no solicitados hasta el desenmascaramiento fue baja (4,6%) y entre los EA locales solicitados el dolor en el sitio de inoculación (2,4%) fue el más común, seguido por el eritema, hinchazón, parestesia, induración y prurito; por otra parte, los EA sistémicos solicitados más usuales fueron la fatiga (8,2%), cefalea (5,9%), mialgia (4%), escalofríos (2,5%), fiebre (1,8%) y diarrea (1,6%), siendo poco comunes la tos, vómitos, erupciones cutáneas y prurito. La eficacia reportada en este ensayo para la prevención de COVID-19 sintomático, 14 días después de la segunda dosis de CoronaVac fue del 83,5% (IC del 95%: 65,4-92,1; p <0,0001), aunque se requiere evaluar los ensayos de fase III, realizados en Brasil, Indonesia y Chile para determinar su eficacia en distintos subgrupos etarios y demográficos 53.
Los datos de seguridad y reactogenicidad de los estudios mencionados, permitieron efectuar un ensayo de fase I (n=72) y fase II (n=480) en individuos de 3 a 17 años, identificando que la mayoría de los EA no serios fueron de leves a moderados y de una duración media de dos días, se resalta que solo dos participantes tuvieron EA de grado 3 en el grupo de la vacuna; no se evidenció EAS relacionados con CoronaVac en esta población. En el análisis según grupos etarios se observó que los EA no serios recopilados durante 28 días posteriores a cada dosis de CoronaVac, tuvieron una mayor prevalencia en participantes de 12 a 17 años (61/161, 37,8%), seguido de grupo de 3 a 5 años (28/113, 24,7%), y de 6 a11 años (20/162, 12,3%) y entre los EA locales y sistémicos solicitados los más frecuentes fueron dolor en lugar de inyección, fiebre, cefalea, tos, y anorexia 54.
En la actualidad existen varias vacunas de virus inactivado en desarrollo clínico de fase III que demuestran, de forma preliminar, resultados satisfactorios sobre seguridad en los ensayos de fase I y II, entre ellas la vacuna de SARS-CoV-2 inactivado desarrollada por el Instituto de Biología Médica y Academia de Ciencias Médicas de China, la vacuna BBIBP-CorV fabricada por el Instituto de Productos Biológicos de Beijing en asociación con Sinopharm y la vacuna de SARS-CoV-2 inactivado producida por el Instituto de Productos Biológicos de Wuhan y Sinopharm 55 - 57.
VACUNAS DE SUBUNIDADES
Las vacunas de subunidades contienen proteínas y dominios proteicos derivadas del SARS-CoV-2, que son capaces de ser reconocidas por componentes moleculares y celulares del sistema inmune, aunque requieren de adyuvantes para inducir una memoria inmune robusta 58. La mayoría de las vacunas basadas en esta plataforma utilizan a la glicoproteína S, dominio S1, S2, RBD y proteínas de la nucleocápside como inmunógenos 59. Las vacunas que se encuentran en ensayo clínico de fase III son la vacuna NVX-CoV2373, basada en glicoproteína S recombinante de SARS-CoV-2, estabilizada en su forma de prefusión y formulada con Matrix-M1, (adyuvante a base de saponina) desarrollada por Novavax y la vacuna ZF2001 que utiliza como antígeno a la forma dimérica de RBD adyuvada con hidróxido de aluminio (Academia de Ciencias de China y Biofarmacéutica Anhui Zhifei Longcom) ( 60 , 61. Ambas vacunas demostraron adecuada seguridad e inmunogenicidad en adultos sanos mayores de 18 años, aunque se requiere evaluar los resultados de los estudios en curso para una conclusión definitiva 61 , 62.
CONCLUSIONES
La pandemia ocasionada por el SARS-CoV-2 es un problema de salud mundial, por lo que es necesario el desarrollo y producción de vacunas para su control. La seguridad y reactogenicidad son aspectos esenciales que se deben considerar para el desarrollo, producción y aprobación de los diferentes candidatos vacunales antes de su administración masiva. La mayoría de los eventos adversos no serios identificados en los diferentes ensayos clínicos fueron leves o moderados, según las especificaciones de las entidades regulatorias, y demuestran una baja incidencia de eventos adversos serios, lo que indica un adecuado perfil de seguridad en adultos mayores de 18 años. La evidencia de algunos estudios sugiere que las vacunas basadas en ARNm contra SARS-CoV-2 son seguras para la población de 12 a 18 años, aunque existen algunos reportes de eventos adversos raros atribuidos a estas vacunas. Actualmente existen escasos reportes acerca de la seguridad de las distintas vacunas contra la COVID-19 en adolescentes y especialmente en niños, por lo que es necesario la conclusión de los distintos estudios en curso y vigilancia poscomercialización para determinar todos los posibles eventos adversos y de especial interés en estas poblaciones.

