










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkHorizonte Médico (Lima)
versión impresa ISSN 1727-558X
Horiz. Med. vol.17 no.3 Lima jul. 2017
http://dx.doi.org/10.24265/horizmed.2017.v17n3.12
CASO CLÍNICO
Diagnóstico molecular de síndrome de Smith-Magenis por MLPA (Multiplex Ligation-dependent Probe Amplification)
Molecular diagnosis of Smith-Magenis syndrome using MLPA (Multiplex Ligationdependent Probe Amplification)
Sergio Talavera Vargas-Machuca 1,a,b, Ismenia Gamboa Oré 1,a,b, Francia Huamán Dianderas c, Ricardo Fujita Alarcónc, María Luisa Fajardo Loo 3, María Luisa Guevara Gil c
1 Biólogo, Docente de la Facultad de Ciencias de la Salud de la Universidad Norbert Wiener.
2 Biólogo, Docente de la Facultad de Medicina de la Universidad Federico Villarreal.
3 Médico Genetista. Instituto Nacional Materno Perinatal. Lima, Perú.
a Laboratorio de Citogenética del Instituto Nacional Materno Perinatal. Lima Perú.
b Docente de la Facultad de Medicina Humana de la Universidad San Martín de Porres.
c Centro de Investigación de Genética y Biología Molecular de la Facultad de Medicina Humana de la Universidad de San Martín de Porres.
RESUMEN
La genética médica avanza rápidamente gracias a las tecnologías que definen con precisión el aporte de los genes en el desarrollo de enfermedades. Algunos síndromes se presentan en la población general, y su diagnóstico y manejo son importantes para brindar al paciente cuidados y pronósticos de vida adecuados. Presentamos el caso de una niña dismórfica nacida a las 33 semanas de gestación por cesárea por preemclampsia materna. El análisis citogenético reveló una deleción heterocigota en el brazo corto del cromosoma 17 (46, XX, del 17p11.2) en el estudio cromosómico. El diagnóstico se complementó con el análisis de MLPA, que mide la presencia/ausencia de ciertos genes definidos en algunos síndromes, y confirmó la deleción de 2.1 megabases que incluyen el gen RAI1, responsable del Síndrome de Smith-Magenis.
Palabras clave: Smith-Magenis; citogenética; MLPA (Fuente: DeSC BIREME).
ABSTRACT
Medical genetics is rapidly advancing thanks to technologies that accurately define which genes are involved in the development of diseases. Some syndromes occur in the general population, and their diagnosis and treatment are important to provide patients with an adequate care and prognosis. We present the case of a dysmorphic child who was born at 33 weeks of pregnancy by caesarean delivery due to preeclampsia. Cytogenetic analysis showed a heterozygous deletion on the short arm of chromosome 17 (46, XX, del 17p11.2). The diagnosis was complemented by MLPA analysis, which measures the presence/absence of certain genes defined in some syndromes, and confirmed the deletion of 2.1 megabases of DNA, including the RAS1 gene, responsible for the Smith-Magenis syndrome.
Keywords: Smith-Magenis; cytogenetics; MLPA (Source: MeSH NLM).
INTRODUCCIÓN
El síndrome de Smith-Magenis (SSM) es una alteración del desarrollo de origen genético, con características físicas que cambian con la edad pero son específicos de la enfermedad. El SSM incluye discapacidad intelectual moderada y algunos signos frecuentes son personalidad ansiosa y agresiva, a veces autoagresión, trastornos de sueño y los síntomas y fallas en el desarrollo tienen una severidad variable (1). Su fascies incluye frente abombada con micrognatia al nacer, que se convierte en prognatismo durante la niñez, ojos hundidos, hipertelorismo, así como nariz y parte media de la cara achatada. Algunos pacientes pueden tener orejas displásicas e implantación baja y posterior, frecuentemente con sordera. Puede haber anomalías esqueléticas con escoliosis, baja estatura, anomalías de las extremidades, de los dedos de pies y manos, así como problemas cardiacos, renales y visuales. Es una enfermedad rara, con una prevalencia estimada de 1:15,000 a 1:25,000 (2) aunque podría ser más frecuente. En general, no se registra casos familiares ni heredados y se cree que la mutación ocurre en las células germinales (formación de espermatozoides u óvulos). El síndrome es causado mayoritariamente (90%) por una deleción de longitud variable en 17p11.2 que incluye varios genes contiguos con el gen RAI1 responsable de la mayor parte de las características, aunque no incluye baja estatura ni problemas auditivos, cardiacos o renales. Se ha descrito que los individuos con mutaciones en RAI1 únicamente, presentan menos anomalías viscerales y talla baja, aunque tienen mayor tendencia a la obesidad (3). Se cree que la variabilidad y severidad depende del número de genes mutados, por ello, determinar los genes involucrados ayudará a caracterizar el fenotipo. Aunque la duplicación de 17p11.2 tiene características similares a SSM esta condición es conocida como Síndrome de Potocki-Lupski (4, 5).
Dependiendo de su tamaño, la detección de un rearreglo genómico puede hacerse a dos niveles: microscópico (citogenético de alta resolución) si el reordenamiento es mayor al límite visible de 2 millones de pares de bases (Mb) y submicroscópico (menor a 2 Mb) para conocer precisamente cuáles son los genes involucrados. Existen muchas afecciones genéticas causadas por rearreglos submicroscópicos como los síndromes de Williams, Angelman/Prader-Willi, Di George y muchas condiciones asociadas a discapacidad intelectual incluyendo SSM (1, 3).
Hay distintas pruebas para detectar reordenamientos genómicos submicroscópicos (deleciones o duplicaciones) de uno o más genes y se basan en la detección de los segmentos correspondientes a los genes que estamos estudiando. Algunas técnicas utilizadas son el Southern blot, el FISH (Fluorescence in situ hybridization) o el dQF-PCR (Quantitative fluorescence polimerase chain reaction) que permiten la identificación de un solo gen (Southern blot), o de algunos genes (FISH o dQF-PCR) que dependen de 2 a 5 fluorocromos. La técnica, llamada MLPA (Multiple Ligation-dependent Probe Amplification), creada por Schouten y col permite la detección (ausencia o presencia) múltiple de hasta 50 genes a la vez (6). Ello se da primero por la presencia de pares de sondas contiguas con secuencias complementarias a los diferentes genes. Si el gen está presente, las dos sondas contiguas van a ligarse y forman un iniciador adecuado para la amplificación por PCR. Contrariamente, si los genes están ausentes, las sondas no encuentran secuencias para localizarse contiguamente y no se forma el iniciador necesario para el PCR (6). Luego de la formación (o no) del iniciador de PCR se procede a la amplificación. El diseño tiene segmentos artificiales que permiten incluir productos múltiples de distinto tamaño adecuándolo a su análisis por longitud. La combinación de electroforesis capilar con láser, permite la detección de hasta 50 distintos amplicones PCR y la cuantificación (0X, 0.5X, 1X, 1.5X etc.) de cada segmento. En el caso de los autosomas, cuando las copias paterna y materna están presentes, se tendrá un valor normal diploide (valor referencial 1X), cuando hay una deleción heterocigota de un gen la sonda muestra un valor referencial de 0.5X. Esta metodología permite el estudio seriado de varios segmentos genómicos simultáneamente, y ha permitido el estudio de enfermedades que involucran la deleción o inserción de muchos genes de forma más económica (Huamán y col, en prep.). En este caso clínico con sospecha de síndrome de Smith Magenis, reportamos el análisis citogenético de una paciente y subsecuentemente la caracterización molecular haciendo uso de la técnica MLPA.
CASO CLÍNICO
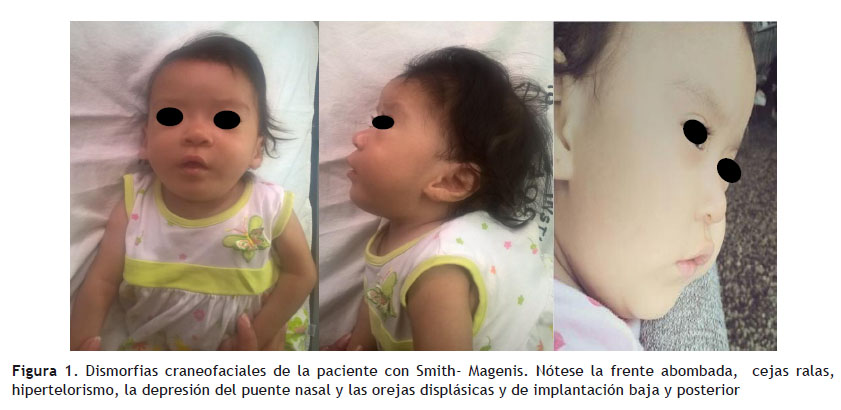
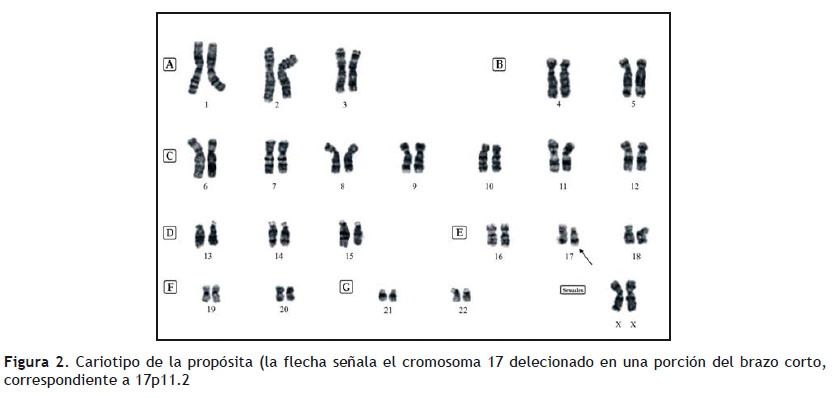
En el Instituto Materno Perinatal (Lima-Perú), nace una niña por cesárea a las 33 semanas de edad gestacional, con 1576 gramos, talla de 38 cm, perímetro cefálico 29.5 cm, y perímetro torácico de 26 cm. El valor de Apgar al primer minuto y a los 5 minutos fue de 8. La madre de 21 años de edad, es ingresada de emergencia por un cuadro de preclampsia severa y refiere haber tenido una pérdida espontánea del primer trimestre en una gestación anterior. El examen físico de la recién nacida revela características craneofaciales dismórficas. La cara es alargada con frente abombada y ojos hundidos, pliegues epicánticos e hipertelorismo, cejas ralas, puente nasal deprimido, orejas displásicas de implantación baja y posterior. Las suturas craneales cabalgadas y el abdomen globuloso (Figura 1). La ecografía transfontanelar establece ventriculomegalia mínima sin hemorragia intraventricular. Los miembros superiores se muestran flexionados con hiperextensión, y las palmas muestran línea simiana, el tercer dedo del pie izquierdo sobresale en altura sobresale el segundo y el cuarto y el tono muscular es ligeramente disminuido. Existe persistencia del conducto arterioso de 1 mm, comunicación interventricular perimembranosa de 2 mm y soplo sistólico II/VI. Además de estos rasgos la niña presenta laringomalacia, úvula bífida y un retraso en el desarrollo psicomotor. La evaluación oftalmológica y la ecografía renal son normales. La niña es referida al Servicio de Genética del mismo Instituto para el examen cromosómico correspondiente. Este se realiza a partir de cultivos de linfocitos obtenidos por punción en sangre periférica en medio de cultivo RPMI 1640. Los cultivos son procesados luego de 72 hs de incubación a 37°C y se utiliza el método de bandeo GTG (Tripsina-Giemsa) en láminas preparadas. La lectura de láminas a una resolución de 550 bandas, permitió identificar un patrón cromosómico constante tipo 46, XX, del 17 (p11.2) en 40 metafases analizadas. Esta alteración corresponde a una pérdida o deleción de un segmento del brazo corto del cromosoma 17 (Figura 2). Con la finalidad de descartar la posibilidad de herencia a partir de algún reordenamiento balanceado que comprometa al cromosoma 17 en alguno de los progenitores, se sugirió el cariotipo a los padres de la niña. El cariotipo de la madre tiene una constitución 46, XX. No fue posible realizar el cariotipo de la niña (Figura 3).

Para la confirmación del diagnóstico citogenético y Facultad de Medicina Humana de la Universidad de San determinación de los genes involucrados se solicitó Martín de Porres. Se hizo la extracción de ADN a partir el estudio de MLPA. Este fue realizado en el Centro de de la muestra de sangre, con el método salting-out Investigación de Genética y Biología Molecular de la modificado en nuestro centro(7). Para el estudio propuesto, se usaron dos mezclas de sondas (3 sondas en SALSA P245 y 7 sondas en SALSA P374, MRC Holland) que corresponden a secuencias nucleotídicas de la región cromosómica 17p11.2 cuyas deleciones causan el síndrome de Smith-Magenis. Se encontró ausencia heterocigota (0.5X) en una región que incluye los genes (de telómero a centrómero) COPS3, MIR33, TOMIL, RAI1, LRRC4, LLGL, PRPSAP y MFAP4 en 17p11.2 indicando la presencia de una deleción de al menos 1 millón pares de bases. Con la SALSA 245 se encuentra la presencia homocigota (1X, normal) de los genes PAFAH1B1 en17p13.3 y NF1 en 17q11.2 y KANSL1 en 17q21.31. Con la SALSA 374 se encuentra la presencia homocigota (1X, normal) de los genes RAP1GAP2 al MNT en 17p13.3. Así, corroboramos la deleción heterocigota de un segmento genómico involucrando varios genes en la región 17p11.2 incluyendo RAI1 y genes contiguos, compatible con la causa molecular del síndrome Smith Magenis.
DISCUSIÓN
Existe una gama de alteraciones cromosómicas y génicas que son responsables de variaciones del fenotipo con diversos grados de desviación del desarrollo normal de una persona. El caso que se discute, muestra la pertinencia de la complementación de técnicas que apoyen al diagnóstico y la consiguiente posibilidad de ofrecer a la familia una orientación más precisa del pronóstico y opciones de tratamiento y seguimiento de un paciente.
En este caso, reportamos el diagnóstico complementario de una deleción de una porción del brazo corto del cromosoma 17 en una niña que presenta alteraciones fenotípicas y del desarrollo global. Si bien la citogenética muestra la deleción de forma clara, es importante precisar los genes que estarían implicados en esta deleción heterocigota. A los rasgos fenotípicos reportados por el médico tratante, se le asocia la posibilidad de un diagnóstico del Síndrome de Smith-Magenis, el cual pudo ser verificado mediante la técnica MLPA. Aquí se confirma la deleción heterocigota de la región 17p11.2 y se identifica los genes (o fragmentos de genes) cuya dosis está a la mitad (0.5X) de lo que correspondería a un fenotipo normal.
El Síndrome de Smith-Magenis (OMIM 182290) según el Centro Nacional de Información en Biotecnología (8), es causado en la mayoría de casos (90%) por una deleción intersticial de 3.7 millones de bases en el brazo corto del cromosoma 17 (17p11.2). Smith- Magenis es llamado "síndrome de genes contiguos" porque abarca segmentos del genoma que involucran más de un gen al igual que el síndrome de Di George o el de Williams (9, 10). Mutaciones en el gen RAI1, dentro de esta región, pueden causar el mismo fenotipo, aunque no están relacionadas a estatura pequeña ni anomalías de las vísceras. Desde la descripción inicial de Smith de 9 pacientes no relacionados, se observó que deleciones de varios genes casi en la totalidad de la banda 17p11.2 son responsables del fenotipo (1, 10). Sin embargo se ha reportado que aún la haploinsuficiencia de únicamente RAI1 sería responsable de cambios de comportamiento, craneofaciales y neurológicos que sufren los pacientes de SSM (11,12 y 13). En cambio algunos cambios cardiacos, renales y talla baja se deberían a otros genes contiguos vecinos a RAI1 delecionados en heterocigosis para un fenotipo normal (12, 13). Un análisis de haplotipos en 14 familias de pacientes con el síndrome y de 6 familias con duplicación de la misma región usando marcadores microsatélites, indica que la deleción y las duplicaciones resultan a partir de crossing-over meióticos que no son recíprocos y que ocurren mediante intercambios inter e intracromosómicos cercanos a las repeticiones que se encuentran en la región. Asimismo, se reportó un punto asequible de recombinación asociado a esa región (11,12,14,15). Es importante considerar la expansión de la deleción porque se han reportado casos en los cuales si ésta es mayor de 4 megabases, se puede presentar un fenotipo que sugiere otro síndrome, similar al de Joubert (OMIM 213300), y que se presenta con ataxia, retraso mental severo, retraso del desarrollo, hipoplasia del vermis cerebelar y patrón anormal de respiración. Si bien no se ha reportado un locus para el síndrome de Joubert en esa región, las anomalías encontradas sugerirían otro locus para este síndrome, además de los ya reportados en otros cromosomas (12,13, 14).
En algunos casos de mutaciones de RAI1 hay problemas para conciliar el sueño, anomalías del comportamiento, piel seca, miopía elevada y comportamiento destructivo que los lleva a la auto-mutilación. Esto se ha comprobado para diferentes tipos de mutaciones que afectan este gen en un análisis retrospectivo de 105 pacientes con el síndrome SSM (8, 13). En otros estudios, Girirajan (8,14) concluyó que el gen principal para la mayoría de rasgos del síndrome de Smith-Magenis es el RAI1, mientras que otros genes dentro de la región 17p11.2 contribuyen con la variabilidad de los rasgos y la severidad del síndrome. De los trabajos de Andrieux (8,15) se verifica que la región delecionada más pequeña que se asocia al síndrome de Smith-Magenis es de 1.4 Mb, que contiene menos de 16 genes y que está ubicada en 17p12-p11.2.
En cuanto a nuestra paciente, la técnica de MLPA muestra que los límites de la deleción están entre los genes COPS3 y MFAP4 que involucran una región de 2.1 megabases según el GRCh38.7 (16). Entre estos genes se encuentran RAI1, algunos relacionados con la formación de purinas y pirimidinas (PRPSAP2) y otro involucrado con la adhesión celular o las interacciones entre células (MFAP4). Se ha reportado una alta actividad del gen RAI1 sobre todo en tejidos neurológicos y se ha observado que en ratones, su expresión es inducida por ácido retinoico (5,14,17).
En conclusión, se reporta el estudio de una paciente con síndrome de Smith-Magenis mediante análisis citogenético y además por MLPA, lo que permite corroborar dicho diagnóstico y además permitirá una evaluación del desarrollo acorde a las manifestaciones clínicas que la paciente presente a lo largo de su vida.
REFERENCIAS BIBLIOGRÁFICAS
1. Smith A, Dykens E, Greenberg F. Behavioral Phenotype of Smith-Magenis Syndrome (del 17p11.2). American Journal of Medical Genetics (Neuropsychiatric Genetics). 1998;81:179– 85. [ Links ]
2. Greenberg F, Guzzetta V, Montes de Oca-Luna R, Magenis R, Smith A, Richter S, et al. Molecular analysis of the Smith– Magenis syndrome: a possible contiguous gene syndrome associated with del(17)(p11.2). Am J Hum Genet. 1991; 49: 1207–18 [ Links ]
3. Gener B. Síndrome de Smith-Magenis. Protoc diagn ter pediatr. 2010;1:37-43 [ Links ]
4. Truong H, Solaymani-Kohal S, Baker K, Girirajan S, Williams S, Vlangos C. Diagnosing Smith-Magenis syndrome and duplication 17p11.2 syndrome by RAI1 gene copy number variation using quantitative real-time PCR. Genet Test. 2008;12(1):67-73. [ Links ]
5. Online Mendelian Inheritance in Man [Internet]. EE. UU: Johns Hopkins University; 2007 [actualizado el 4 de octubre del 2012; acceso 28 de mayo del 2017]. POTOCKI-LUPSKI SYNDROME. Disponible en: http://omim.org/entry/610883 [ Links ]
6. Schouten J, McElgunn C, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30(12):e57 [ Links ]
7. Miller S, Dykes D, Polesky H. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research. 1988; 16(3): 1215 [ Links ]
8. Online Mendelian Inheritance in Man [Internet]. EE. UU: Johns Hopkins University; 1987 [actualizado el 4 de octubre del 2012; acceso 28 de mayo del 2017]. Smith-Magenis Syndrome. Disponible en: http://omim.org/entry/182290 [ Links ]
9. Myers S, Challman T. Congenital heart defects associated with Smith-Magenis syndrome: two cases of total anomalous pulmonary venous return. Am J Med Genet A. 2004;131(1):99- 100. [ Links ]
10. Slager R, Newton T, Vlangos C, Finucane B, Elsea S. Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet. 2003;33(4):466-8. [ Links ]
11. Shaw C, Bi W, Lupski J. Genetic proof of unequal meiotic crossovers in reciprocal deletion and duplication of 17p11.2. Am J Hum Genet. 2002;71(5):1072-81 [ Links ]
12. Bi W, Park SS, Shaw C, Withers M, Patel P, Lupski J. Reciprocal crossovers and a positional preference for strand exchange in recombination events resulting in deletion or duplication of chromosome 17p11.2. Am J Hum Genet. 2003;73(6):1302-15 [ Links ]
13. Edelman E, Girirajan S, Finucane B, Patel P, Lupski J, Smith A, Elsea S. Gender, genotype, and phenotype differences in Smith-Magenis syndrome: a meta-analysis of 105 cases. Clin Genet. 2007;71(6): 540-50. [ Links ]
14. Girirajan S, Vlangos C, Szomju B, Edelman E, Trevors C, Dupuis L, et al. Genotype-phenotype correlation in Smith-Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum. Genet Med. 2006; 8: 417-27 [ Links ]
15. Andrieux J, Villenet C, Quief S, Lignon S, Geffroy S, Roumier C, et al. Genotype-phenotype correlation of 30 patients with Smith-Magenis syndrome (SMS) using comparative genome hybridisation array: cleft palate in SMS is associated with larger deletions. J Med Genet. 2007; 44(8): 537-40 [ Links ]
16. The National Center for Biotechnology Information [Internet]. EE. UU.; 2017 [acceso 28 de mayo del 2017]. Assembly: GRCh38. Disponible en: https://www.ncbi.nlm.nih.gov/ assembly?term=GRCh38&cmd [ Links ]
17. Lucas R, Vlangos C, Das P, Patel P, Elsea S. Genomic organisation of the ~1.5 Mb Smith-Magenis syndrome critical interval: transcription map, genomic contig, and candidate gene analysis. Europ J Hum Genet. 2001; 9(12): 892-902. [ Links ]
Fuentes de financiamiento:
Este artículo ha sido financiado por los autores.
Conflictos de interés:
Los autores declaran no presentar ningún conflicto de interés.
Correspondencia:
Sergio Hugo Talavera Vargas Machuca Dirección: Jr. Miro Quesada 941 Cercado de Lima. Lima, Perú. Teléfono: 991568878
Correo electrónico: stalavera35@hotmail.com
Recibido: 20 de febrero de 2017
Evaluado: 28 de febrero de 2017
Aprobado: 08 de junio de 2017

