










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkHorizonte Médico (Lima)
Print version ISSN 1727-558X
Horiz. Med. vol.18 no.3 Lima July/Set. 2018
http://dx.doi.org/10.24265/horizmed.2018.v18n3.13
CASO CLÍNICO
Síndrome de Jeune en un paciente pediátrico: Reporte de caso clínico
Jeune syndrome in a pediatric patient: A clinical case report
Elert Zapata Aguilar 1,a ORCID iD: https://orcid.org/0000-0002-3805-099X, Víctor Hugo Calderón Arenas 1,a ORCID iD: https://orcid.org/0000-0002-9470-0248, Lizeth Y. Cabanillas Burgos 1,b ORCID iD: https://orcid.org/0000-0003-1072-9981
1. Hospital Base Carlos Alberto Seguín Escobedo. EsSalud. Arequipa, Perú.
a. Médico Asistente. Servicio de UCI Pediátrica.
b. Médico Residente. Servicio de UCI Pediátrica.
RESUMEN
El síndrome de Jeune es una displasia esquelética de baja frecuencia, de transmisión autosómica recesiva y potencialmente mortal. Clínicamente tiene una amplia variedad de manifestaciones; En forma clásica se presenta con un tórax estrecho, hipoplasia pulmonar secundaria, braquimelia, anormalidades pélvicas, cardiacas y renales. El compromiso pulmonar es variable y puede ser letal. El diagnóstico puede sospecharse prenatalmente con el estudio ultrasonográfico al medir la circunferencia torácica y de la jaula costal, estudiar su relación entre sí y con la circunferencia abdominal. En este artículo se describe el caso clínico de una paciente de 3 meses de edad, con forma de presentación leve y diagnóstico posnatal.
Palabras clave: Enfermedades del recién nacido; Displasias óseas; Enfermedades hereditarias (Fuente: DeCS BIREME).
ABSTRACT
Jeune syndrome is a low frequency, autosomal recessive and potentially life-threatening skeletal dysplasia. From a clinical perspective, it has a wide variety of manifestations; however, it is commonly characterized by a narrow thorax; secondary pulmonary hypoplasia; brachymelia; and pelvic, cardiac and renal abnormalities. Lung involvement is variable and may be lethal. The diagnosis may be suspected prenatally through an ultrasonography, by measuring the thoracic and rib cage circumference, and studying the relation between them and with the abdominal circumference. This article describes the clinical case of a 3-month-old female patient with a mild disease presentation and postnatal diagnosis.
Keywords: Neonatal diseases; Bone dysplasia; Genetic diseases (Source: MeSH NLM).
INTRODUCCIÓN
El síndrome de Jeune (SJ), conocido también como distrofia torácica asfixiante es una enfermedad autosómica recesiva rara causada por una alteración genética localizada en el cromosoma 15q13 (1,2). La incidencia en los Estados Unidos es de un caso por cada 100 000 a 130 000 nacidos vivos (2). Este síndrome se distingue por una displasia ósea con distintas alteraciones en tórax, páncreas, corazón, riñón y retina (3). El cuadro presenta una policondrodistrofia caracterizada por costillas anchas, cortas, horizontales y uniones costocondrales irregulares, con una caja torácica rígida y estrecha (3,4) que es extremadamente reducida y causa una severa restricción pulmonar (5). La edad de presentación clínica está correlacionada con la severidad de la enfermedad; presenta una gran variabilidad en el cuadro clínico por lo que se puede clasificar como letal, severo, moderado y forma latente (5). En el período perinatal ocurre la mayoría de las muertes, debido al tórax estrecho y a los pulmones hipoplásicos que conllevan a la asfixia (2,6). En sus formas moderadas, el niño presenta neumonías de repetición con insuficiencia respiratoria progresiva en el primer año de vida. Por regla general, esos pacientes son dependientes de la ventilación mecánica(7,8). Algunos pacientes diagnosticados más tarde pueden presentar ventilación adecuada en reposo y mejorar la capacidad respiratoria con el pasar de los años. La función pulmonar varía desde normal o puede llegar a la hipertensión pulmonar a causa de la hipoplasia pulmonar(1,5,7). El compromiso cardiaco ocasional proviene de la insuficiencia cardiaca secundaria y del aumento de la resistencia vascular pulmonar, de la constricción torácica, de la hipoplasia alveolar y de la enfermedad miocárdica primaria (1,3). Puede haber un compromiso hepático con ictericia neonatal prolongada, enfermedad poliquística hepática, hiperplasia de los ductos biliares y cirrosis hepática congénita (2-4). Los pacientes pueden tener una mala absorción intestinal, degeneración de retina, polidactilia y anormalidades dentarias y pélvicas. Además de eso, presentan insuficiencia renal que se debe a la fibrosis intersticial difusa con infiltración linfoblástica y alteraciones tubulares (dilatación y atrofia intercaladas) (9). La poliuria, polidipsia y la hipertensión pueden estar presentes en el segundo o tercer año de vida, mientras que el fracaso renal se desarrolla en la infancia o en la adolescencia (1,9).
El objetivo de este artículo es reportar el caso de una lactante mujer con síndrome de Jeune tipo II, oligoasintomática, sin otras malformaciones mayores asociadas; quien presentó un cuadro de neumonía a los 3 meses de edad que evolucionó favorablemente con tratamiento médico, no siendo tributaria de ventilación mecánica, lo cual contrasta con las presentaciones habituales: formas severas y letales.
CASO CLÍNICO
Se describe el caso clínico de una lactante de 3 meses de edad, producto de segundo embarazo, gestación doble; padres no consanguíneos, madre de 33 años de edad sin antecedentes de complicaciones durante la gestación. La paciente nació por cesárea segmentaria a las 37 semanas de gestación, Apgar 7 al minuto y 8 a los 5 minutos; peso al nacer 2900 g, longitud al nacer 48 cm, perímetro cefálico 36,5 cm, perímetro torácico: 29 cm, presentó al nacimiento taquipnea y saturación baja. Al examen físico se evidenció estrechez de la caja torácica, extremidades cortas, manos y pies pequeñas; no requirió ventilación a presión positiva ni medidas de reanimación, pero si oxigenoterapia suplementaria. Permaneció hospitalizada en la Unidad de Neonatología del Hospital Carlos Alberto Seguín durante 14 días por taquipnea transitoria del recién nacido. La ecocardiografía que se le realizó mostró una comunicación interauricular (CIA) de 0,3 milímetros. Evolucionó favorablemente siendo dada de alta a los 15 días de vida. A los 3 meses de edad ingresó a la Unidad de Cuidados Intensivos Pediátricos del Hospital Carlos Alberto Seguín por presentar dificultad respiratoria, sensación de alza térmica y tos. A su ingreso tuvo un peso de 4900 gramos, una longitud de 50 centímetros, perímetro torácico de 32 centímetros, FC: 158 latidos por minuto, FR: 65 respiraciones por minuto, SatO2: 83 %. En el examen se observó tórax asimétrico, estrecho, polipneico, retracciones subcostales e intercostales moderadas, se auscultó disminución del murmullo vesicular en ambos hemitórax y crepitantes bilaterales a predominio de base de hemitórax derecho, abdomen globuloso, respiración abdominal; a nivel de extremidades se observó extremidades cortas y braquimelia (Figura 1).
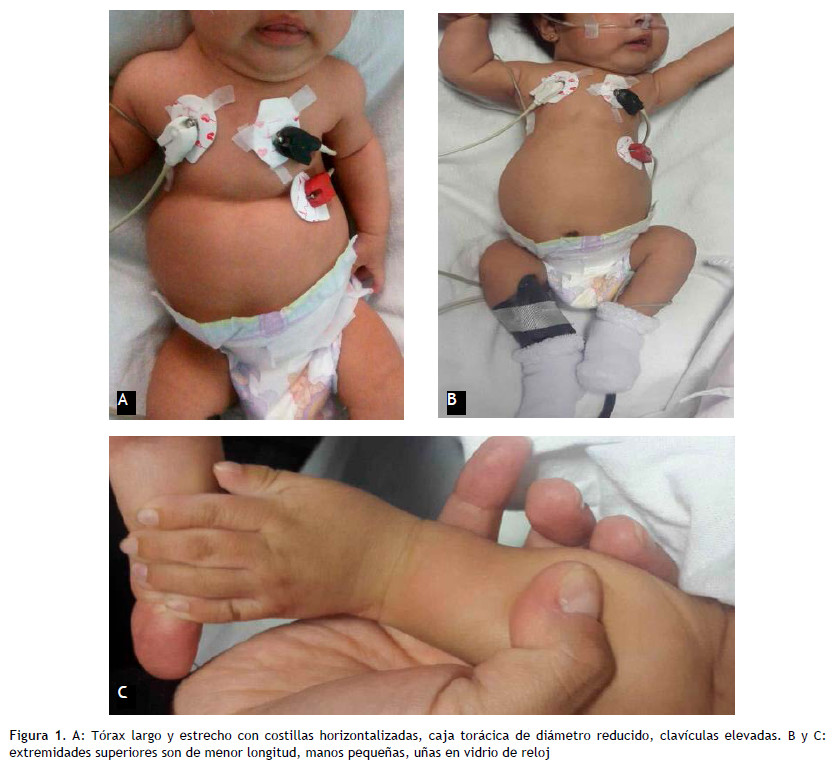
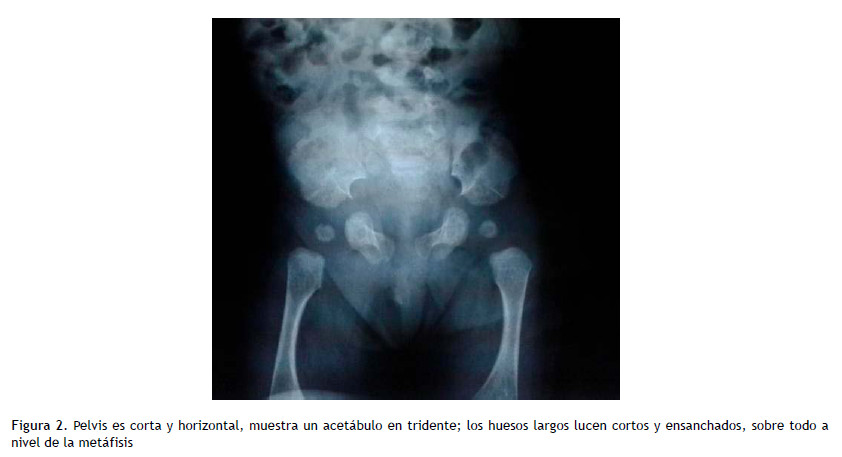
Se realizó biometría hemática, presentando una hemoglobina de 14,5 g/dl, Hgma 13 300/mm3, A: 0 %, S: 63 %, L: 28 %, M: 7 %, E: 2 %. Plaquetas: 611 000/ mm3. Creatinina: 0,37 mg/dl, glucosa en 80 mg/dl. AGA y e- arterial con pH: 7,42, pO2: 111 mmHg, pCO2: 34 mmHg, HCO3: 22,1 mEq/l. Na: 135 mEq/l, K: 4,3 mEq/l. La radiografía de tórax mostró radiopacidad bilateral perihiliar sugestiva de compromiso parenquimal e intersticial sin derrame pleural; por lo que se inició soporte oxigenatorio y antibióticos por vía endovenosa (ceftriaxona). A las 24 horas de iniciado el tratamiento, la gasometría de control evidenció pH: 7,37, pO2: 70 mmHg, pCO2: 50 mmHg, HCO3: 28,9 mEq/l, Na: 132 mEq/l, K: 5,4 mEq/l. Ante la sospecha de displasia torácica (síndrome de Jeune) y patologías asociadas, se le realizó una ecocardiografía que fue informada como situs solitus, concordancia aurículoventricular, CIA tipo ostium secundum de 0,3 mm con shunt de izquierda a derecha, no se encontró signos de hipertensión pulmonar. La ecografía abdominal y renal descartó malformaciones asociadas. Se realizó radiografía de caderas en la que se evidenció la forma en tridente del acetábulo (Figura 2).
Durante su hospitalización presentó episodios de regurgitaciones constantes. La gammagrafía con DPTATC99M evidenció reflujo gastroesofágico con pico dominante a los 19 minutos hasta tercio medio y ausencia gammagráfica de microaspiraciones, recibiendo tratamiento con omeprazol vía oral. Evolucionó favorablemente con el tratamiento antibiótico endovenoso y la oxigenoterapia siendo dada de alta a los 10 días de su ingreso, sin necesidad de oxígeno domiciliario, con buena tolerancia oral y ganancia adecuada de peso. Se le programó además controles ambulatorios por las especialidades de neumología, gastroenterología y cardiología pediátrica.
DISCUSIÓN
En 1955, Jeune y colaboradores describieron dos hermanos enfermos de lo que ellos denominaron "Distrofia torácica asfixiante de carácter familiar" o displasia torácica asfixiante (DTA) (1-5). Desde esa época, hasta la fecha, se han reportado más de 100 casos, predominantemente en publicaciones europeas especializadas en radiología. El síndrome de Jeune se hereda de forma autosómica recesiva. Puede deberse a un defecto congénito del metabolismo por una alteración en la formación endocondral del hueso. La base molecular del síndrome ha sido en parte develada, afectando a los genes IFT80 (3q25.33), DYNC2H1 (11q22.3), WDR19 (4p14) and TTC21B (2q24.3). Cada uno codifica para una proteína de transporte intraflagelar, lo que confirma que el síndrome de Jeune pertenece al grupo de ciliopatías. Su incidencia se calcula en 1 a 1,4 casos por cada 100 mil a 130 mil nacidos vivos (2). No existe predilección por condición étnica ni por sexo. Se puede expresar clínicamente de dos formas. La primera se trata de una forma neonatal potencialmente letal, la cual, a su vez se divide en dos subtipos: la de pacientes que requieren una inmediata ventilación mecánica y no se pueden destetar, y la de aquellos que después de varios brotes de neumonía recurrente requieren intubación y soporte ventilatorio. Desde el punto de vista mecánico, estos pacientes presentan enfermedad pulmonar restrictiva severa; con pulmones severamente hipoplásicos y frecuentemente presentan hipertensión pulmonar. La segunda forma, de menor gravedad, se presenta en niños que sobreviven a la etapa neonatal, los cuales presentan una pared torácica estrecha (10); con síntomas respiratorios moderados al nacer y pueden no necesitar ventilación mecánica en años (11).
La piedra angular que definirá la evolución y pronóstico será el compromiso pulmonar debido a la restricción que ejerce la caja torácica sobre los pulmones, lo que determina una disminución de los movimientos respiratorios torácicos con una respiración casi exclusivamente diafragmática y abdominal (12).
Las diversas fuerzas que actúan sobre la estructura mecánica de la pared torácica desempeñan un papel importante en la determinación del volumen pulmonar, y las anomalías de la pared torácica pueden tener un impacto significativo en la función pulmonar; en particular, la patología de la pared torácica puede ser un importante contribuyente a la fisiología restrictiva del sistema respiratorio. La caja torácica y la columna vertebral forman los límites de la pared torácica superior y lateralmente, mientras que el diafragma constituye el componente inferior. El límite externo de la pared torácica inferior es el borde inferior de la caja torácica, pero internamente, este límite se solapa con el diafragma, que se encuentra adyacente a la mitad de la caja torácica. En virtud de esta "zona de aposición" entre el diafragma y las costillas, la caja torácica está expuesta tanto a presiones abdominales como pleurales, de modo que la función normal de la pared torácica depende de la intrincada interacción de los dos espacios. Los cambios en el contenido intraabdominal o el incremento de la pared abdominal alteran las propiedades mecánicas de la pared torácica a través de sus efectos sobre el movimiento del diafragma (13,14).
La mayoría de las anomalías de la pared torácica se asocian con una disminución de la compliance del sistema respiratorio. Esto resulta en una disminución en la capacidad pulmonar total, disminuyendo la capacidad residual funcional y el volumen residual.
Las pruebas de función pulmonar permiten determinar el grado de insuficiencia respiratoria causada por una anormalidad de la pared torácica y si otras enfermedades respiratorias están contribuyendo a los síntomas del paciente (15). Los pacientes con defectos ventilatorios restrictivos más severos están en riesgo de hipoventilación e hipercapnia. El punto en el que se desarrolla la hipercapnia varía entre los pacientes según el tipo de anormalidad de la pared torácica y otros factores que afectan la compliance de la pared torácica y la fuerza de los músculos respiratorios.
La clasificación del síndrome de Jeune se basa en sus dos formas clínicas de presentación mencionadas anteriormente: la forma mayor o tipo I y la forma menor o tipo II. El tipo I se observa en el 70 % de los casos y corresponde a pacientes que presentan un tórax pequeño, rígido y estrecho por las alteraciones costales características de la enfermedad, con respiración fundamentalmente abdominal y sintomatología respiratoria precoz y grave (insuficiencia respiratoria severa de presentación en la etapa neonatal). Estos niños terminan requiriendo ventilación mecánica a las pocas horas de nacer y, por lo general con parámetros altos (FiO2 >= 60 %, PIM>=35 PEEP>=8). Presentan severas complicaciones respiratorias y una alta incidencia de mortalidad antes del año de vida(3,5,9).
El tipo II se presenta en el 30 % de los casos. Se observa en pacientes con malformaciones costales intermedias, escasa o nula sintomatología y sobrevida prolongada. En el seguimiento a largo plazo se observa una notoria mejoría de los estigmas radiológicos y la configuración torácica levemente alterada va normalizándose de forma progresiva (3,5,9).
El diagnóstico del síndrome de Jeune se establece por las características clínicas y las alteraciones esqueléticas. Las costillas son cortas y horizontales, y dan por resultado un tórax fijo, con una caja torácica estrecha y expansión de las uniones costocondrales. Las clavículas son "altas" en la radiografía de tórax. Frecuentemente, como en el caso que se presenta, la pelvis es corta y horizontal y muestra un acetábulo en tridente; los huesos largos lucen cortos y ensanchados, sobre todo a nivel de la metáfisis; las falanges son hipoplásicos, con epífisis terminadas en forma de cono. Usualmente presentan estatura corta o enanismo (16). El diagnóstico es usualmente confirmado por la demostración radiológica de las malformaciones esqueléticas típicas, como son un tórax estrecho, acortamiento de las extremidades, polidactilia y anormalidades pélvicas. Estos hallazgos le han dado a esta enfermedad la denominación alternativa de distrofia torácica-pélvica-falángica. La tomografía computarizada de emisión (TAC) de tórax con reconstrucción tridimensional de la pared también puede objetivar todas estas alteraciones. Para algunos autores, como Jacquier, la medición del perímetro torácico determinaría el pronóstico de estos pacientes, un perímetro torácico inferior a 28 cm es de muy mal pronóstico (17).
Se ha informado sobre la detección de fetos afectados en el segundo y tercer trimestre de familias con alto riesgo; en la ecografía prenatal podemos encontrar los hallazgos característicos. Otros hallazgos ecográficos incluyen polihidramnios y movimientos respiratorios fetales ausentes o débiles. Las pruebas de función pulmonar pueden revelar enfermedad pulmonar restrictiva grave. La biopsia renal puede revelar una displasia tubular quística con o sin esclerosis glomerular. La prioridad en el manejo de pacientes con SJ es apoyar la función respiratoria.
Se ha observado también distrofia retiniana que puede afectar la visión. La frecuencia real de estas complicaciones es difícil de estimar, ya que los casos más benignos y menos complicados frecuentemente no se reportan en la literatura (12,14,15). Otras alteraciones asociadas incluyen polidactilia preaxial y postaxial, uñas hipoplásicas, anormalidades dentales y lengua lobulada.
Los hallazgos anatomopatológicos en necropsias revelan que a nivel de las uniones condrocostales se encontrará osificación endocondral que progresa de manera desordenada y sería la causa determinante de la disminución de la longitud costal. A nivel pulmonar, se detectó hipoplasia pulmonar de grado variable, seguramente secundaria a la estrechez de la caja torácica ya que en todos los casos el desarrollo bronquial es normal, pero presentan disminución de las divisiones alveolares.
En el diagnóstico diferencial de esta patología, existe un importante número de síndromes genéticos que presentan costillas cortas y anchas; sin embargo, la de mayor interés por sus semejanzas es la displasia condroectodérmica o síndrome de Ellis-Van Creveld.
La forma de presentación menor habitualmente no requiere tratamiento. Estos pacientes pueden ser asintomáticos o presentar síntomas respiratorios leves. En el seguimiento a largo plazo se observa una notoria mejoría de los estigmas radiológicos. La configuración torácica, levemente alterada, progresivamente adquiere características normales (7,10).
En la forma mayor, aparte del tratamiento ventilatorio de sostén, se puede considerar el tratamiento quirúrgico con el fin de expandir la caja torácica para lograr un aumento de la capacidad respiratoria (16). Sin embargo, es importante determinar el momento oportuno para la cirugía. La corrección quirúrgica debería realizarse en forma precoz y no esperar a que el paciente permanezca en respiración mecánica por tiempo prolongado por las complicaciones que esta genera, que condicionan una evolución tórpida y un aumento de la morbilidad y mortalidad en estos niños (17).
En los últimos años varios procedimientos quirúrgicos se han desarrollado para corregir la anomalía torácica asociadas, todos son objeto de polémica en la edad neonatal. El objetivo de la cirugía es facilitar el aumento del volumen pulmonar, aunque el éxito del tratamiento depende del nivel de hipoplasia pulmonar que se tenga (17,18).
En este artículo se reporta el caso de una lactante mujer con síndrome de Jeune, cuya forma de presentación fue del tipo menor, oligoasintomática, sin otras malformaciones mayores asociadas; que presentó un cuadro de neumonía a los 3 meses de edad con evolución favorable con tratamiento médico y que no requirió ventilación mecánica, la cual contrasta con las presentaciones habituales, formas severas y letales. En este caso correspondería al síndrome de Jeune, tipo II, de quien se espera una buena evolución, pero que puede tener ciertas limitaciones futuras que implicarían un manejo multidisciplinario a posteriori.
REFERENCIAS BIBLIOGRÁFICAS
1. Chen H. Asphyxiating thoracic dystrophy (Jeune Syndrome). eMedicine, 2009. [ Links ]
2. N Morgan, C Bacchelli, P Gissen, J Morton, G Ferrero, M Silengo, et al. A lócus for asphyxiating thoracic dystrophy, ATD, maps to chromosome 15q13. J Med Genet. 2003;40(6):431-435. [ Links ]
3. Acastello E, Garrido P. Actualización de la clasificación de las malformaciones congénitas de la pared torácica: 22 años de experiencia en un hospital pediátrico. Rev Med Clin Condes. 2009;20(6): 758-767. [ Links ]
4. Orfaliais CS, March MFP, Ferreira S, Alves R, Sant’Anna CC. Distrofia torácica asfixiante de Jeune: relato de 3 casos. J Pediatr. 1998; 74(4):333-337.
5. Schwartzstein RM. Chest wall diseases and restrictive physiology. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc.2018 [ Links ]
6. Díaz ZS. Displasia torácica asfixiante o síndrome de Jeune. Rev Chil Obstet Ginecol. 2003; 68(4): 322-6. [ Links ]
7. Do Ngoc TC, Barois A, Estournet MB, Bataille J, Ioos C. Jeune’disease (asphyxiating thoracic dystrophy) and respiratory failure: importance of early respiratory management with periodic hyperinsufflation. Bull Acad Natl Med. 2007; 191(7): 1411-29.
8. Rodriguez Molinero L, ]imenez Mena E, Muro Tudelilla LM, Gonzalez Perez A, Rodriguez Sanchez C, Fernandez de las Heras F. Distrofia torácica asfixiante o enfermedad de Jeune. Bol Pediatr. 1990; 31:135-139.
9. Niaudet P. Clinical manifestations, diagnosis, and treatment of nephronophthisis. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc.2018. [ Links ]
10. Elcioglu NH, Hall CM. Diagnostic dilemmas in the short ribpolydactyly syndrome group. Am J Med Genet. 2002; 111(4): 392-400. [ Links ]
11. Casteels I, Demandt E, Legius E. Visual loss as the presenting sign of Jeune syndrome. Eur J Paediatr Neurol. 2000; 4(5): 243-7. [ Links ]
12. Jankauskiene A, Bernatoniene J. Clinical quiz. Jeune syndrome. Pediatr Nephrol. 2000; 14: 1054-6. [ Links ]
13. Zanelli SA. Asphyxiating Thoracic Dystrophy (Jeune Syndrome). Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc.2018. [ Links ]
14. Sahin N, Kara H, Ertugrul F, Aydogdu T, E İcel. Jeune Sendromu ve Anestezi. Turkiye Klinikleri. J Anest Reanim. 2007; 5(3):150- 153.
15. Borland LM. Anesthesia for chilren with Jeune’s Syndrome (asphyxiating thoracic dystrophy). Anesthesiology. 1987; 66(1):86-88.
16. Schinzel A, Savoldelli G, Briner J, Schubiger G. Prenatal sonographic diagnosis of Jeune Syndrome. Radiology. 1985:154 (3):777-778. [ Links ]
17. Davis JT, Ruberg RL, Leppink DM, McCoy KS, Wright CC. Lateral thoracic expansion for Jeune’s asphyxiatin dystrophy: a new approach. Ann Thorac Surg. 1995; 60(3):694-696.
18. Davis JT, Heistein JB, Castile RG, Adler B, Mutabagani KH, Villalobos RE, et al. Lateral thoracic expansion for Jeune’s syndrome: midterm results. Ann Thorac Surg. 2001; 72(3):872-877.
Fuentes de financiamiento:
Este artículo ha sido financiado por los autores.
Conflictos de interés:
Los autores declaran no tener ningún conflicto de interés.
Correspondencia: Lizeth Y. Cabanillas Burgos
Dirección: Urbanización Las Buganvillas R 14, Cerro Colorado.
Arequipa, Perú.
Teléfono: 054 987316719 / 054 445701
Correo electrónico: lyzyu_6@hotmail.com
Recibido: 16 de abril de 2018.
Evaluado: 23 de mayo de 2018.
Aprobado: 08 de junio de 2018.

