











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Los organismos multicelulares utilizan diversas formas de señalización celular para controlar los mecanismos de crecimiento, migración, supervivencia y diferenciación. Además, involucran una gran cantidad de ligandos (como proteínas, péptidos y ciertos lípidos) que se unen a receptores, los cuales propician cambios conformacionales que causan reacciones en cascada de diversas proteínas (transducción de señales) las que, frecuentemente, convergen en algunas vías intracelulares bien conservadas y están vinculadas a la regulación transcripcional de los programas de genes (1). Una de las vías más ampliamente estudiada en las últimas décadas ha sido la vía PI3K/AKT/mTOR, cuyos componentes participan en una amplia gama de procesos de regulación celular como crecimiento, proliferación, apoptosis, metabolismo, migración, inmunidad y secreción celular. Además, las aberraciones en la señalización de PI3K/AKT/mTOR contribuyen a un espectro igualmente amplio de enfermedades humanas como cáncer, trastornos inmunológicos, trastornos neurológicos, diabetes, sobrecrecimiento de tejidos localizados y enfermedades cardiovasculares 2,3,4.
El cáncer involucra una serie de alteraciones genéticas y epigenéticas que se van adquiriendo a través del tiempo mediante múltiples pasos. No todas las mutaciones genómicas afectan la función celular (mutaciones pasajeras), pero ciertas mutaciones son clave para el desarrollo del cáncer y generan ventajas para el crecimiento selectivo (mutaciones conductoras). Dentro de este contexto, las señales de proliferación continua están asociadas al concepto de oncogenes, los cuales son genes conductores mutados que favorecen la activación celular o ganancia de funciones. En contraparte, existen los genes supresores de tumores que, por mutaciones en ambos alelos de estos genes conductores, inactivan las funciones de proteínas supresoras, importantes en el control celular. La mayoría de los cánceres son causados por dos y hasta ocho alteraciones conductoras que pueden desarrollarse, a veces, a través de décadas 4,5.
Los genes conductores más frecuentemente mutados corresponden a los genes KRAS (16,5 %) y PIK3CA (13,6 %); sin embargo, cuando se evalúa la tasa de mutación en las distintas vías de señalización, existe una correlación con lo mencionado, ya que el 27,9 % de los tumores más frecuentes tuvieron alguna mutación en la vía MAPK; mientras que 17,6 % correspondieron a la vía PI3K/AKT/mTOR. Los cánceres con mayor tasa de mutación en esta última vía son cáncer de mama, colon y endometrio 6,7,8.
Por tanto, la PI3K/AKT/mTOR es una de las vías intracelulares más importantes asociadas al cáncer, lo cual ha llevado al desarrollo de innumerables estudios moleculares y enormes esfuerzos para la síntesis de medicamentos dirigidos a esta vía de señalización. Hoy, algunos de estos medicamentos se emplean en las distintas fases de los ensayos clínicos, y cada vez es más claro que los inhibidores de PI3K y mTOR son efectivos para detener la progresión tumoral 9.
El objetivo de esta revisión sistemática cualitativa es conocer más profundamente el funcionamiento de la vía PI3K/AKT/mTOR, sus elementos participantes, activación, supresión, alteraciones más importantes en el desarrollo del cáncer y principales tipos de cáncer implicados y, de esa manera, tener una visión cercana de su funcionamiento y sus alteraciones.
ESTRATEGIA DE BÚSQUEDA
Las bases de datos consultadas fueron PubMed y Scopus, en el periodo comprendido entre el 1 de enero de 2000 y diciembre de 2020. Las siguientes palabras clave utilizadas se utilizaron en la búsqueda: Fosfatidilinositol 3-Quinasa, Proteínas Proto-Oncogénicas c-akt y Serina- Treonina Quinasas TOR asociadas a cáncer. De los artículos seleccionados, se excluyeron las revisiones repetitivas, ya que las fuentes originales fueron consideradas prioritarias. Fueron incluidas las enfermedades más importantes ligadas a las alteraciones de la vía de señalización PI3K: carcinoma de mama, carcinoma de tiroides, glioblastoma multiforme, carcinoma endometrial, cáncer de próstata y linfomas.
FOSFOLÍPIDOS DE LA MEMBRANA CELULAR: FOSFATIDIL INOSITOL
Los fosfoglicéridos o glicerofosfolípidos son los principales constituyentes lipídicos de la membrana celular. Están compuestos de una molécula de glicerol, en la que se han esterificado dos ácidos grasos y un grupo fosfato unido a un alcohol o un aminoalcohol. Una fracción baja de los fosfoglicéridos corresponde a los fosfolípidos de inositol, los cuales se concentran en la superficie citosólica de membranas y se sintetizan en el retículo endoplásmico, principalmente. Más que una función estructural, los fosfolípidos de la membrana celular tienen importantes roles en la señalización celular 10.
El fosfatidilinositol (PI) puede fosforilarse a través de cinasas específicas y de forma selectiva en las posiciones 3, 4 y 5 para generar siete especies únicas, de las cuales tres son monofosfatos; tres, bifosfatos, y un trifosfato 11. Los monómeros son el fosfatidilinositol 3 fosfato (PI3P), asociado con la formación de membranas endosómicas y con la preformación de autofagosomas, presumiblemente en el retículo endoplásmico12; el fosfatidilinositol 4 fosfato (PI4P), aún sin un rol claro en regulación celular, pero que es un substrato importante en la formación de fosfatidilinositol 4,5 bifosfato 13; y el fosfatidilinositol 5 fosfato (PI5P), que tiene funciones regulatorias 14. Los bifosfatos son los siguientes: el fosfatidilinositol 4,5 bifosfato [PI(4,5)P2], intermediario esencial que, a través de la estimulación de un receptor, activa la fosfolipasa C (PLC) que hidroliza PI(4,5)P2 para generar diacilglicerol (DAG) e inositol-1,4,5-trisfosfato (IP3), los cuales inician así una cascada de señalización en relación a la liberación de calcio del retículo endoplásmico 15; el fosfatidilinositol 3,5 bifosfato [PI(3,5)P2], que no ha sido suficientemente estudiado, pero tendría injerencia en la homeostasis celular y en la adaptación16; el fosfatidilinositol 3,4 bifosfato [PI(3,4)P2], que parece tener un papel significativo en la migración celular mediante la unión a reguladores citoesqueléticos específicos17. Finalmente, el fosfatidilinositol 3,4,5 trifosfato [PI(3,4,5) P3], que sería el más importante en relación a su función celular (Figura 1).
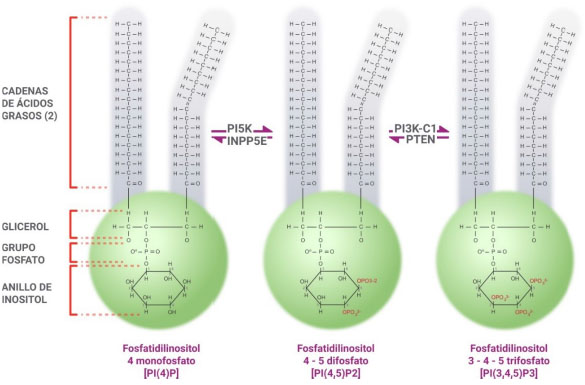
El fosfatidilinositol 4 monofosfato es transformado en fosfatidil inositol 4,5 difosfato o PIP2 a través de la enzima fosfoinositol 5 quinasa (PI5K) y es revertida por la fosfatasa Inositol polifosfato 5-fosfatasa E (INNPP5E). PIP2 es convertido a PIP3 por la enzima fosfatidilinositol 3 quinasa C1 (PI3K-C1). La fosfatidilinositol-3,4,5-trisfosfato 3-fosfatasa (PTEN) revierte esta reacción, lo que la hace un punto de regulación muy importante.
ACTIVACIÓN DE PI3K
Existe un grupo de enzimas de la familia fosfatidilinositol 3 quinasa (PI3K), cuyas funciones son fosforilar el grupo hidroxilo de la posición 3' del anillo de inositol en la membrana plasmática y generar segundos mensajeros importantes tales como fosfatidilinositol-3,4-bifosfato [PI(3,4)P2] y fosfatidilinositol-3,4,5-trifosfato [PI(3,4,5)P3]. Conocemos actualmente tres diferentes clases de PI3K, que a su vez se dividen en subclases según su afinidad por algunos sustratos, su homología y las funciones que cumplen 18. La clase I es la mejor caracterizada en cuanto a su implicancia en la patogénesis del cáncer y sus PI3K se dividen en subclase IA y IB. La clase II incluye tres enzimas diferentes (PI3K-C2α, PI3K-C2β y PI3K-C2γ) y aún es un enigma, a pesar de los estudios recientes que brindan pistas novedosas sobre su papel en la transducción de señales. Finalmente, solo hay un miembro conocido de clase III (PI3K-C3 o Vps34), que está implicado en la respuesta celular a los cambios nutricionales 19.
PI3K clase IA consiste de una de tres isoformas catalíticas (p110 a, b, y d) codificadas por los genes PIK3CA, PIK3CB, y PIK3CD, y una subunidad reguladora, que puede ser p85α o sus variantes por splicing (p55α y p50α), p85β o p55g (Figura 2).
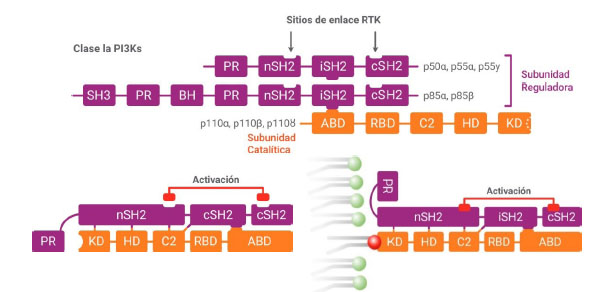
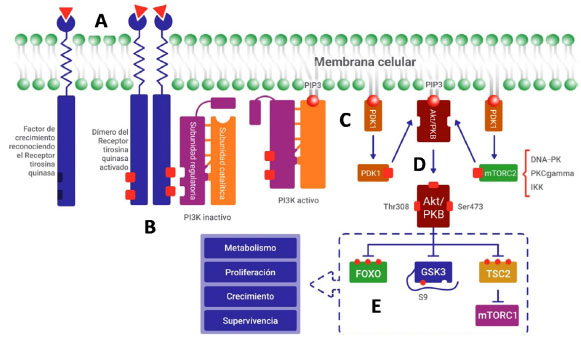
En condiciones fisiológicas, PI3K se activa mediante señales extracelulares como factores de crecimiento (factor de crecimiento epidérmico, factor de crecimiento derivado de plaquetas, factor de crecimiento similar a la insulina), citoquinas, hormonas, entre otros, que interactúan con un receptor tirosina quinasa (RTK), lo cual induce a su dimerización y producción de cambios conformacionales a partir de la fosforilación de los residuos de tirosina en las regiones citoplasmáticas del RTK (Figura 3A). Estos cambios en el dominio citoplasmático del RTK permiten el
reclutamiento de proteínas adaptadoras (IRS1-2, SHC, GRB2 y GAB2), las cuales activan la proteína RAS, o directamente a PI3K, lo que resulta, al final, en la activación alostérica de PI3K 20,21. A nivel molecular, la fosforilación de la subunidad p85 en forma directa o a través de los intermediarios permite un cambio conformacional de la molécula de PI3K que expone el dominio con actividad catalítica de la proteína p110; en consecuencia, un grupo fosfato se agrega al fosfatidilinositol 4,5-bisfosfato (PIP2) y lo transforma en fosfatidilinositol 3,4,5 trifosfato (PIP3) 22 (Figuras 3B y 3C).
ACTIVACIÓN DE AKT
Entre las quinasas convencionales, la familia AGC (derivado de las proteínas quinasas A, G y C) es un subtipo de quinasas serina/treoninas específicas ligadas a las proteínas G, dentro de las cuales están la proteína quinasa B (PKB), mejor conocida como Akt, algunos de los sustratos de mTORC2, quinasas inducidas por suero/glucocorticoides (SGK) y la proteína quinasa C (PKC). Akt es fundamental en la regulación del metabolismo, supervivencia, motilidad, transcripción y progresión del ciclo celular 23.
La producción de PI(3-4)P2 y PIP3 es esencial para el reclutamiento y activación de la proteína Akt derivada de la activación del RTK. Hay tres isoformas de Akt, cada una compuesta por un dominio de homología de Pleckstring (PH) en el extremo N-terminal, un dominio quinasa interno y un dominio regulador C-terminal. El dominio PH aislado de cada isoforma de Akt puede unirse al PI(3-4)P2 o PIP3 con afinidad similar, pero en el contexto de la proteína completa, Akt1 y 3 se unen preferentemente a PIP3, mientras Akt2 se une preferentemente a PI(3-4)P2(24). De cualquier manera, esta unión provoca el reclutamiento de Akt en la membrana celular, donde se les unirá otra serina/treonina quinasa que contiene el dominio PH, conocida como quinasa 1 dependiente de 3-fosfoinositol (PDK1), la cual fosforila AKT en el residuo de treonina 308 (Thr308) y produce un aumento sustancial en la actividad de Akt 25. Sin embargo, para una activación completa, AKT necesita ser fosforilada en un residuo de serina en posición 473 (Ser473), en particular por mTORC2 (26), pero se ha visto que también puede fosforilarse por otras quinasas como DNA-PK 27, PKCγ 28, IKK 29 o por autofosforilación de AKT 30 (Figura 3D).
La Akt fosforilada actúa activando o inhibiendo diferentes proteínas ubicadas en la membrana plasmática, citosol o en el núcleo celular, de tal forma que existen más de 100 proteínas de numerosas clases funcionales reportadas en la literatura, que incluyen quinasas proteicas y lipídicas, factores de transcripción, reguladores de proteínas G pequeñas y del tráfico de vesículas, enzimas metabólicas, ubiquitina ligasas E3, reguladores del ciclo celular y muchos otros. Akt fosforila estas proteínas a nivel de los residuos serina y treonina, dentro de regiones de reconocimiento específicas, para activar o, lo más frecuente, inhibir la función de la proteína objetivo 31. Muchos de los sustratos fosforilados por mediación de AKT tienen una única consecuencia fisiológica; sin embargo, AKT también controla los nodos de señalización clave que luego regulan múltiples objetivos y funciones celulares 32 (Figura 3E). Las principales funciones de Akt son las siguientes: aumenta el transporte de GLUT4 a la membrana celular para permitir el ingreso de glucosa; incrementa la síntesis de proteínas y bloquea la autofagia (a través de mTOR1); induce la glucógeno sintetasa y, por tanto, la síntesis de glucógeno (por inhibición del GSK3); aumenta la proliferación celular (al inhibir la proteína supresora FOXO), y promueve la apoptosis (al actuar con otras proteínas importantes como BAD, MDM2, CHK1). Por ello, inferimos que un error genético en estos elementos tiene serias consecuencias metabólicas y aumentan las posibilidades de oncogénesis.
Glucógeno sintasa quinasa 3 (GSK3)
La GSK3 es una proteína quinasa de serina/treonina que fosforila zonas específicas de proteínas en los residuos de aminoácidos de serina y treonina. Tiene dos isoformas, GSK-3a (51 kDa) y GSK-3b (47 kDa), que están codificadas por distintos genes y a menudo tienen funciones superpuestas. Se describió inicialmente como una enzima clave involucrada en el metabolismo del glucógeno, sin embargo, ahora se sabe que regula una amplia gama de funciones celulares. La GSK3 tiene las características de una quinasa no convencional: está constitutivamente activa, sus sustratos necesitan una fosforilación previa por otra quinasa, y se inhibe en lugar de activarse como respuesta a la estimulación de las dos de sus principales vías de señalización: insulina y proteínas Wnt. Ha sido relacionada con enfermedades como la diabetes mellitus tipo 2, enfermedad de Alzheimer, trastornos bipolares, inflamación, entre otros 33.
La mayoría de los sustratos de GSK-3 descritos hasta el momento contienen una región tipo S/T-x-x-x-S/T (inicia con serina o treonina, seguido de tres aminoácidos cualquiera y la cuarta posición cierra con S/T); usualmente el prefosforilado se produce en la última posición. Ello supone que los sustratos de GSK3 siempre están regulados por un mecanismo que implica dos quinasas. El grupo fosfato del sustrato interactúa con GSK3 en un bolsillo formado por tres aminoácidos de carga positiva (R96, R180 y K205) 34. La acción inactivadora del AKT sobre el GSK3 se produce cuando en el extremo N-terminal, la activación de la proteína quinasa B (PKB), fosforila un residuo de serina (el residuo Ser-9 en GSK3β y Ser-21 en GSK3α) e inhibe la actividad de GSK-3, que resulta en la ocupación del bolsillo cargado positivamente, por la parte N-terminal de la proteína, que debe plegarse para conseguir esta acción, lo cual oculta la región inicial del S/T-x-x-x-S/T y no permite la segunda fosforilación, inactivando GSK3 35. La actividad completa de GSK-3 requiere no solo de la ausencia de la fosforilación en el extremo N-terminal, sino también la presencia de una fosforilación en posición Y279 en GSK-3a. Sin embargo, esta fosforilación activadora de tirosina de GSK-3 ocurre por autofosforilación 36.
Factores de transcripción FOXO
Las proteínas FOXO son un subgrupo familiar de factores de transcripción Forkhead que llegan a más de 100 miembros sobre la base de similitud de secuencia. Estas proteínas participan en muy diversas funciones: así, los miembros de la clase "O" provienen de la vía de señalización de insulina/PI3K/Akt. Akt y las quinasas inducidas por suero/ glucocorticoides (SGK) fosforilan directamente los factores FOXO en tres residuos conservados, ello produce el secuestro citoplasmático de FOXO y, por tanto, represión de su actividad transcripcional a nivel del núcleo celular. Además de ser fosforilado por AKT y SGK, FOXO también puede tener modificaciones postraduccionales en muchos otros residuos que pueden contribuir a la regulación de expresión génica, sobre todo en el contexto del metabolismo/resistencia al estrés, en respuesta a diversos estímulos externos 37,38.
FOXO tiene un papel funcional en procesos como la regulación del ciclo celular, muerte celular, metabolismo, protección contra el estrés por oxidación y supervivencia. El hecho de que muchas proteínas de Forkhead actúan como efectores terminales de varias vías de señalización clave apoya la hipótesis de que pueden actuar como integradores moleculares de señales extracelulares que permitirían la conversación cruzada entre vías de señalización paralelas y, por lo tanto, respuestas más eficientes y adecuadas a las fluctuaciones ambientales. El papel clave de los factores de algunos Forkhead en el desarrollo embrionario se evidencia por las consecuencias de las mutaciones y/o desregulaciones encontradas en once genes FOX humanos, relacionadas con enfermedades genéticas humanas 39,40.
Complejo de la esclerosis tuberosa (TSC)
El complejo TSC consiste en la proteína hamartina (codificada por el gen TSC1) y la proteína tuberina (gen TSC2), las cuales no muestran secuencias homólogas con otras proteínas conocidas. El complejo TSC1 y TSC2 agrupa a proteínas traducidas de genes supresores de tumores, cuyas mutaciones conducen a la esclerosis tuberosa, un trastorno genético que afecta al 0,01-0,02 % de todos los recién nacidos y se caracteriza por la formación de tumores benignos en la piel, cerebro, corazón, riñones y muchos otros órganos 41. TSC1/2 es un regulador crítico de mTORC 1. TSC1 (hamartina) estabiliza TSC2 (tuberina), que es la proteína activadora de GTPasa (GAP) que actúa sobre Rheb (homólogo de Ras enriquecido en el cerebro) en la regulación de mTORC1. Las mutaciones de la línea germinal en TSC1 o TSC2 provocan el desarrollo de tumores benignos (hamartomas) debido a la señalización hiperactiva de mTORC1, que, por lo general, no produce malignidad, con algunas excepciones 42.
mTOR (MECHANICIST TARGET OF RAPAMICINA)
La proteína mTOR es una quinasa serina/treonina relacionada con la vía de PI3K, que forma una subunidad catalítica en dos complejos proteicos distintos, conocidos como complejos mTOR 1 y 2 (mTOR C1-C2). El complejo mTORC1 se define por sus tres componentes principales: mTOR, Raptor (proteína reguladora asociado con mTOR) y mLST8. Raptor facilita el reclutamiento del sustrato para mTORC1 a través de la unión a la proteína de señalización TOR (TOS); y mLST8 se asocia con el dominio catalítico de mTORC1. Además de estos tres componentes principales, mTORC1 también contiene las dos subunidades inhibidoras PRAS40 (sustrato de Akt rico en prolina de 40 kDa) y DEPTOR, una proteína de interacción mTOR. La estructura de mTORC2 también contiene mTOR y mLST8, pero, en vez de Raptor, mTORC2 contiene Rictor (compañero de mTOR insensible a rapamicina), una proteína que probablemente cumple una función análoga; mTORC2 también contiene DEPTOR, así como las subunidades regulatorias mSin1 y Protor 1/2. La rapamicina se une a la proteína citosólica específica FKPB-12: este complejo inhibe la activación de mTOR1. El complejo rapamicina-FKBP12 no se une o inhibe directamente a mTORC2; sin embargo, el tratamiento prolongado con rapamicina anula la señalización de mTORC2, probablemente por la incapacidad de mTOR unido a rapamicina de incorporar nuevos complejos a mTORC2 43,44.
Los estudios han demostrado que mTOR está involucrada en muchas vías de señalización en el cuerpo que incluyen PI3K/AKT, subunidad 1 y 2 del complejo de esclerosis tuberosa (TSC1-2)/Rheb, LKBL/AMPK. La principal función de mTORC1 es metabólica, ya que esta vía integra entradas de, al menos, cinco señales extracelulares e intracelulares importantes (factores de crecimiento, estrés, estado de la energía, oxígeno y aminoácidos para controlar muchos procesos importantes, incluyendo la síntesis de proteínas y lípidos y autofagia) para promover el crecimiento celular cuando la energía es suficiente y promover el catabolismo cuando se ingresa al ayuno. Por su parte, mTORC2 controla, principalmente, la proliferación celular, estructura citoesquelética y supervivencia 45. El regulador natural de mTORC1 es el heterodímero TSC1/ 2, cuya estimulación provoca la formación de una proteína activadora de GTPasa (GAP), la cual es una enzima que hidroliza la molécula guanosina trifosfato de Rheb (homólogo del RAS enriquecido en el cerebro) que, cuando está activo, estimula la función de mTORC1. Por tanto, el complejo TSC es, finalmente, un inhibidor (supresor tumoral) 46.
ALTERACIONES GENÉTICAS DEL PI3K EN CÁNCER
La mutación oncogénica de la isoforma catalítica PI3K p110α es la más frecuente en los cánceres humanos, mientras que las isoformas catalíticas p110β, p110δ y p110γ rara vez están mutadas, pero pueden estar sobreexpresadas; la mutación o pérdida de expresión de la isoforma reguladora p85α también está asociada con cáncer. Aunque las isoformas catalíticas de la PI3K clase IA comparten similitudes estructurales y de sustrato, tienen papeles distintos en la mediación de la señalización de PI3K en diferentes contextos fisiológicos y oncogénicos. Las células cancerosas con mutaciones de los receptores de tirosina quinasas, mutaciones oncogénicas de RAS, dependen en gran medida de p110α, incluso en presencia de mutación o pérdida de PTEN 47.
La mayoría de las mutaciones ocurren en dos de los dominios de p110a: helicoidal y quinasa. Dentro del dominio helicoidal, los puntos calientes más comunes son Glu542 y Glu545, los cuales, por lo general, mutan a lisina y se encuentran en la interfaz con el dominio nSH2 de la subunidad p85. Parece ser que el contacto entre estos aminoácidos y el dominio nSH2 se produce dentro de una región del dominio nSH2 que también está en contacto con el dominio quinasa de p110; de esta forma, las mutaciones del dominio helicoidal pueden alterar la actividad de la enzima. En el dominio de la quinasa, His1047 está frecuentemente mutada a arginina dentro de una hélice al final del bucle de activación. Otra mutación quinasa menos frecuente es Met1043, la cual ocurre dentro de la misma hélice y es probable que tenga efectos similares sobre la actividad enzimática. Las mutaciones Thr1025 se encuentran cerca del extremo N del bucle catalítico, lo que puede alterar directamente la conformación del bucle catalítico como mecanismo por el cual la mutación altera la actividad enzimática (48). Recientemente, se ha demostrado que la pérdida parcial de la subunidad p85α aumenta la unión de los complejos de señalización p110α-p85 a los receptores activados, lo cual aumenta la activación de la vía PI3K e implica un papel para la p85 libre monomérica como un regulador negativo de la señalización de PI3K (49) (Figura 4).
Las mutaciones oncogénicas de la subunidad p110α son las más frecuentes en los cánceres humanos; existen también mutaciones de la isoforma reguladora p85α asociadas con cáncer. Las mutaciones más frecuentes en los dominios correspondientes están pintadas de anaranjado oscuro. ∆ Ex11: cambios en el exón 11; en las siguientes, la letra mayúscula inicial corresponde al aminoácido original; el número, a la posición del aminoácido dentro de la estructura primaria de la proteína, y la letra mayúscula final, al aminoácido reemplazante por la mutación. PR: dominio rico en prolina; SH2: homólogo 2 de la proteína Src; nSH2, cSH2 e iSH2 (dominios SH2 N- y C-terminal e inter SH2; KD: dominio quinasa; HD: dominio helical; C2: dominio C2; RBD: dominio de ligando; ABD: dominio del ligando adaptador.
La activación de la señalización de PI3K está regulada negativamente por tres clases de fosfatasas de inositol polifosfato. La fosfatasa del fosfoinositol 3-fosfato o PTEN desfosforila la posición 3 de PI3,4,5 P3 para generar PI(4,5)P2; mientras que la fosfatasa del fosfoinositol 5-fosfato como la proteína homologa Src-2 que contiene inositol 5-fosfatasa (SHIP) y la fosfatasa de la fosfatidilinositol 4,5-bisfosfato 5-fosfatasa (PIB5PA) desfosforilan la posición 5 para producir PI3,4,5P2.
Esta última se somete a su vez a la desfosforilación por polifosfato de inositol 4-fosfatasa tipo I (INPP4A) y tipo II (INPP4B) en la posición 4 para generar PI(3)P, terminando así la señalización de PI3K (50) (Figura 1).
La proteína inhibitoria, fosfatasa y homóloga de tensina (PTEN), que puede eliminar un fosfato de la molécula de PIP3 para convertirlo en PIP2, es el regulador negativo más importante de esta vía y uno de los supresores tumorales con el mayor efecto en diferentes tipos de cáncer 51. La mutación de ambos alelos de PTEN en las células germinales ha sido ampliamente evidenciada en la enfermedad de Cowden, la cual también se conoce como síndrome de hamartoma múltiple: los pacientes tienen predisposición al cáncer con un patrón de herencia autosómico dominante y alta susceptibilidad al carcinoma de mama y, en menor medida, al carcinoma tiroideo 52. Existen mutaciones somáticas del PTEN principalmente en el glioblastoma y carcinoma endometrial, y en menor proporción en cáncer de próstata, linfomas y cáncer de mama 53.
Los niveles de PIP3 también pueden ser regulados por otro supresor tumoral conocido como polifosfato de inositol 4-fosfatasa tipo II B (INPP4B), que tiene el mismo efecto que PTEN. El INPP4B actúa como un supresor tumoral en varios cánceres epiteliales al inhibir la señalización de PI3K/Akt. La pérdida de INPP4B ha sido considerada un marcador de agresividad en algunos cánceres, como los de mama triple negativos 54; paradójicamente, el INPP4B puede tener un comportamiento contrario a lo esperado, ya que también muestra características promotoras de tumores en leucemias y cáncer de mama 55.
PRINCIPALES ALTERACIONES GENÉTICAS DE AKT EN CÁNCER
Las mutaciones en AKT1-3 ocurren en 3 a 5 % de los cánceres humanos, pero la mayoría de ellas son mutaciones pasajeras 56. Se ha identificado un grupo de amplificaciones o mutaciones activadoras en los genes AKT en múltiples tumores sólidos, aunque con frecuencias mucho más bajas que los activadores e inhibidores de PIK3CA. La mutación activadora más conocida es la mutación E17K en el dominio PH de AKT1 en pacientes con cáncer de mama, especialmente los subtipos luminales, cáncer de ovario y colon. Esta mutación confiere una activación constitutiva de la quinasa; sin embargo, E17K es incapaz de promover la génesis tumoral sin contar con otras mutaciones conductoras 57,58.
En oposición a lo descrito, Kim buscó la presencia de la mutación E17K en los genes AKT1 2 y 3 en 731 tejidos cancerosos: se detectaron cuatro mutaciones AKT1 E17K en los cánceres de mama (4/93; 4,3 %), pero ninguna en otros cánceres. No hubo mutación en los genes AKT2 o AKT3. Este estudio demostró que la mutación AKT1 E17K ocurre en los cánceres de mama en baja frecuencia, y que es rara en otros cánceres comunes, incluidos los carcinomas colorrectales, pulmonares, gástricos y hepatocelulares y las leucemias agudas. A pesar de la función oncogénica confirmada del AKT1 E17K, las incidencias raras de la mutación sugieren que puede no desempeñar un papel crucial en el desarrollo de los tipos más comunes de cánceres humanos 59.
Glucógeno sintasa quinasa 3 (GSK3) en cáncer
GSK-3 se sobre expresa en diversas afecciones cancerosas, como tumores de colon, hígado, ovario y páncreas. La disminución de GSK-3β inhibe el crecimiento del cáncer de páncreas, la angiogénesis y la expresión del factor de crecimiento endotelial vascular. Sin embargo, el papel de GSK-3 en el cáncer a menudo depende de mTOR, una molécula de señalización crucial en la proliferación celular 60.
Factores de transcripción FOXO en cáncer
La fusión de FOXO1 humano (conocido como FKHR; forkhead en rabdomiosarcoma) con la proteína PAX 3/7 en el rabdomiosarcoma alveolar (tumor pediátrico del músculo esquelético) es el resultado de la translocación cromosómica t (2; 13) (q35; q14) y t (1; 13) (p36; q14), que provoca la formación de la mayoría de los rabdomiosarcomas alveolares altamente metastásicos y agresivos 61,62. Estos cambios genéticos son somáticos y no se heredan en la familia. De manera similar, la translocación cromosómica de FOXO3 (también conocido como FKHRL1) y FOXO4 (también conocido como AFX) con el gen MLL produce una leucemia de linaje mixto, un cáncer hematológico agresivo en niños, caracterizado por la presencia de una proteína de fusión MLL-FOXO3/4, resultante de la translocación cromosómica t (6; 11) (q21; q23) y t (X; 11) (q13.1; q23) 63,64. Es importante destacar que estos reordenamientos genéticos en el rabdomiosarcoma alveolar y en la leucemia de linaje mixto no solo crean nuevas proteínas de fusión oncogénicas, sino que también causan la pérdida del alelo FOXO del cromosoma original, lo que resulta en haploinsuficiencia del locus FOXO. El gen FOXO3 es un supresor tumoral casi siempre eliminado durante la carcinogénesis de adenocarcinoma de pulmón en estadio temprano 65,66. Existen otras entidades oncológicas raras en las cuales los genes FOXO participan, sin embargo, estos genes rara vez mutan en los cánceres humanos, con las excepciones antes descritas.
Complejo de la esclerosis tuberosa (TSC) en cáncer Las mutaciones de la línea germinal en TSC1 y TSC2 predisponen a esclerosis tuberosa, que se caracteriza por el desarrollo de hamartomas generalizados en varios órganos; no obstante, la pérdida de la función de TSC1 o TSC2 rara vez se encuentra en tumores malignos. Como contradicción, la presencia de TSC1/TSC2 en los tumores humanos sugiere que retener cierta actividad supresora del crecimiento podría ser beneficioso durante la evolución del tumor, tal vez al promover la supervivencia cuando las células crecen en un ambiente limitado de nutrientes 67. Existen algunas excepciones conocidas de alteraciones genéticas en TSC y cáncer, como mutaciones somáticas de TSC1 en neoplasia de vejiga 68 o TSC2 en el carcinoma hepatocelular 69.
ALTERACIONES GENÉTICAS DE mTOR EN CÁNCER
La activación de la señalización de mTOR en varios tipos de tumores malignos humanos es un hecho frecuente, pero no se han reportado mutaciones en estos genes supresores tumorales. La activación constitutiva de la señalización de PI3K-mTORC1 en las células cancerosas inhibe fuertemente la autofagia, la cual parece tener un comportamiento contradictorio en la carcinogénesis, ya que actúa como un supresor tumoral en unos casos y como protector de la supervivencia de las células cancerosas en otros. Por tanto, el rol de la autofagia activada por mTORC1 sobre las células cancerígenas depende de un contexto específico: al inicio, es importante para prevenir el cáncer, pero es requerido por el tumor para proteger sus células cuando ya está establecido 70.
Se sabe que la actividad de mTORC2 es alta en las líneas celulares del glioma y otros tumores primarios, en comparación con el tejido cerebral normal, y sugiere que mTORC2 promueve la proliferación de células tumorales y el potencial invasivo debido a la mayor formación de complejos en presencia de una sobre expresión de la proteína Rictor 71.
ALTERNATIVAS TERAPEÚTICAS DE LA VIA PI3K/AKT/ mTOR EN EL CÁNCER
Es probable que la vía PI3K/AKT/mTOR sea una de las más importantes en la visión oncológica dada la alta frecuencia de desregulación de sus componentes encontrada en los casos de cáncer y, por tanto, una de las más investigadas. Se han evaluado más de cuarenta compuestos dirigidos a esta vía en diferentes tipos de cánceres humanos en distintas fases clínicas de investigación, aunque muy pocos han llegado a fases III/IV, debido a la toxicidad que generaban estos productos.
Entre los compuestos que han llegado al mercado existen tres inhibidores de mTOR aprobados por la Food and Drug Administration (FDA) en Estados Unidos: sirolimus, everolimus y temsirolimus con propiedades inmunosupresoras y antiproliferativas, por lo cual se usan en pacientes trasplantados, esclerosis tuberosa, psoriasis y algunos tipos de cáncer (riñón, mama) 72. En el Perú se comercializan bajo las mismas indicaciones aprobadas por la FDA.
La FDA ha aprobado cinco inhibidores de PI3K entre los años 2014y2021que son los siguientes: idelalisib (inhibidordePI3K delta), copanlisib (inhibidor de PI3K, predominantemente contra PI3K-α y PI3K-δ), duvelisib (un inhibidor dual oral de PI3K-delta y PI3K-gamma) y umbralisib (inhibidor oral de PI3K-delta y CK1-epsilon). Todos ellos aprobados para la leucemia linfocítica crónica y linfoma folicular en pacientes que han recibido al menos dos terapias sistémicas previas por recurrencia o progresión de enfermedad; el quinto inhibidor, idelalisib (un inhibidor específico de PI3K alfa), es el único inhibidor de PI3K aprobado por la FDA (mayo de 2019), en combinación con fulvestrant, para el tratamiento de mujeres postmenopáusicas y de varones con cáncer de mama avanzado o metastásico con receptores hormonales positivos (RH+), receptor tipo 2 del factor de crecimiento epidérmico humano negativo (HER2-) y presencia de mutaciones en el gen PIK3CA, después de la progresión de enfermedad con o luego de un régimen de tratamiento que incluyó terapia endócrina 73,74,75. En el Perú, todavía no se cuenta con estos medicamentos.
Se están investigando nuevos compuestos con mayor potencia y selectividad, pero se necesitan biomarcadores predictivos que ayuden a tomar las decisiones terapéuticas 76. Hasta el momento, no ha sido posible entender completamente la vía de señalización PI3K/AKT/mTOR debido a su compleja estructura: 254 componentes con 478 enlaces entre ellos, que regulan 17 procesos celulares en total 77.
CONCLUSIONES
La vía PI3K/AKT/mTOR es muy importante en la regulación celular de muchos procesos fundamentales para la célula: crecimiento, proliferación, apoptosis, metabolismo, entre otros, cuyas alteraciones conllevan a una amplia gama de enfermedades humanas y, dentro de ellas, el cáncer.
La activación del fosfoinositol trifosfato (PIP3) ocurre a través de los receptores tirosina quinasa (como los receptores de insulina o del factor de crecimiento epidermal humano), que son puntos referenciales de alteraciones oncogénicas y, por tanto, fuentes de investigación para nuevos medicamentos anticancerígenos. Las alteraciones más profundas de la célula no han mostrado una frecuencia importante, como uno imaginaría, respecto al cáncer.
La vía de señalización PI3K/AKT/mTOR es muy compleja, con muchos componentes e interacciones, y regulan cerca de 17 procesos celulares importantes.
Aunque muchos productos con distintos mecanismos de acción sobre la vía PI3K han sido desarrollados y llevados a la experiencia clínica, solo algunos compuestos han recibido la aprobación regulatoria: los inhibidores de mTOR en cáncer de riñón y mama, inhibidores no selectivos de PI3K en leucemias crónicas y linfomas foliculares, y el inhibidor selectivo alpelisib para el tratamiento del cáncer de mama avanzado/metastásico en segunda línea (junto con el antiestrogénico fulvestrant) en mujeres con la mutación en la subunidad catalítica alfa de la fosfatitilinositol-4,5-bifosfato 3-quinasa. En el Perú, aún no contamos con los inhibidores de PI3K.
Las referencias bibliográficas sobre esta vía de señalización en Latinoamérica, y particularmente en el Perú, son escasas, por ello es necesario entender claramente la función tan importante de esta vía en el desarrollo de diferentes enfermedades e iniciar investigaciones básicas que contribuyan a su conocimiento.

