










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkRevista Peruana de Biología
versión On-line ISSN 1727-9933
Rev. peru biol. vol.19 no.3 Lima dic. 2012
TRABAJOS ORIGINALES
Variabilidad genética y distribución geográfica del maní, Arachis hypogaea L. en la Región Ucayali, Perú
Genetic variability and geographic distribution of peanut Arachis hypogaea L. in Ucayali, Peru
Luis Fernando Rimachi 1, D. Andrade 1, Milusqui Verástegui 1, Jaime Mori 4, Victor Soto 1, Rolando Estrada J.2,3
1 Instituto Nacional de Innovación Agraria- INIA. Av. La Molina # 1981, Apartado Postal 2791, La Molina, Lima Perú.
2 Sub Dirección de Recursos Genéticos y Biotecnología (SUDIRGEB), Instituto Nacional de Innovación Agraria (INIA). Av. La Molina 1981, La Molina, Lima Perú.
3 Facultad de Ciencias Biológicas , Universidad Nacional Mayor de San Marcos, Ciudad Universitaria, Av. Venezuela s/n.
4 Universidad Arzobispo Loayza, Lima, Perú.
Resumen
El Perú ha sido reconocido como uno de los más importantes centros de diversidad del cultivo de maní y de acuerdo a la evidencia arqueológica, pudo haber sido el centro de origen para dicho cultivo. Para incrementar el conocimiento de la diversidad genética del maní en el Perú, se evaluaron 65 accesiones de maní, correspondientes a 21 variedades locales, de las cuencas de los ríos San Alejandro, Ucayali y Aguaytía, de la Región Ucayali. Las accesiones fueron proporcionadas por el proyecto "Modelos de diversidad y de erosión genética en cultivos tradicionales: Asesoría rápida y detección temprana de riesgos usando herramientas SIG", ejecutado en el Instituto Nacional de Innovación Agraria. Se utilizó la técnica AFLP para estimar la variabilidad genética del cultivo, así como para identificar áreas con la mayor riqueza genética. Se obtuvo un total de 157 bandas polimórficas (45,6%), a partir de 10 combinaciones de iniciadores AFLP en nuestras 65 entradas de maní. Se consideraron sólo 135 bandas polimórficas, en base a su contenido de información polimórfica (0,1<PIC< 0,5), para los análisis de similaridad genética y agrupamiento. Se conformaron 8 grupos principales a un nivel de similitud 0,65 en el dendrograma de ligamiento completo, los cuales fueron evaluados según su Correlación, Reproducibilidad y Estructura. El programa de cómputo DIVA-GIS y los datos de pasaporte de las accesiones, junto a los marcadores AFLP obtenidos, identificaron a la cuenca del río Ucayali como el área geográfica con la mayor cantidad de grupos genéticos de maní.
Palabras clave: Biodiversidad; marcadores AFLP; diversidad genética; mani cultivado.
Abstract
Peru has been recognized as one of the most important centers of diversity of the peanut crop and according to archaeological evidence, may have been the center of origin for it. Due to poor knowledge of the current levels of genetic diversity of peanut in Peru, they were evaluated 65 peanut accessions, corresponding to 21 local varieties from the basins of the rivers San Alejandro, Ucayali and Aguaytia, in the Ucayali region; kindly provided by the project "Models of diversity and genetic erosion of traditional crops: Rapid advice and early detection of risks using GIS tools", performed at the "Instituto Nacional de Innovacion Agraria". AFLP technique was used to estimate the genetic variability of the crop in the region and to identify areas with the greatest genetic wealth. There were a total of 157 polymorphic bands (45.6%), from 10 AFLP primer combinations in our 65 entries of peanuts. We considered only 135 polymorphic bands, based on their polymorphic information content (0.1 <PIC <0.5), for analysis of genetic similarity and grouping. Eight groups were formed leading to a similar level of 0.65 in the complete linkage dendrogram, which were evaluated by correlation, reproducibility and structure. The computer program DIVA-GIS and passport data of accessions with AFLP markers, identified the Ucayali river basin as the geographical area with the greatest amount of genetic groups of peanuts.
Keywords: Biodiversity; AFLP markers; genetic diversity; Cultivated peanut.
Introducción
El maní (Arachis hypogaea L.) es la tercer leguminosa de importancia mundial, originaria de Sudamérica, donde se reconoce al Perú como centro de diversificación genética (Stalker & Chapman 1989). Esta especie fue ampliamente cultivada por los nativos del nuevo mundo en el tiempo de la expansión europea por el siglo XVI y fue llevado a Europa, África, Asia e Islas del Pacífico. Los primeros restos arqueológicos del maní cuentan con una antigüedad de 2000 años ac. y han sido hallados en el Perú, fuera de su hábitat silvestre (Sauer 1993).
Es probable que la especie Arachis hypogaea L. se halla originado en el sur de Bolivia y noreste de Argentina (Krapovickas 1969) sobre los 25° S. Las condiciones climáticas y la topografía de ésta región se encuentra entre las más variables del mundo. En esta región existen especies silvestres emparentadas con el maní, como la especie tetraploide A. monticola Krapov. & Rigoni, considerada como el prototipo del maní y biosistemáticamente una forma silvestre de Arachis hypogaea L. (Singh & Moss 1982).
Aunque no hay suficientes datos para establecer cuándo ocurrió la domesticación del maní, existe evidencia arqueológica que sugiere que la domesticación del maní fue anterior al del maíz de Huaca Prieta. El maní no está representado en los restos pre-cerámicos, pero parece haber sido introducido en asociación con las primeras cerámicas. Los datos de carbono de este período, y por lo tanto del maní, fluctúan entre los 1200 a 1500 años ac. (Hammons 1973). Evidencia arqueológica indica gran variación en los manís hallados en Supe, ciudad costeña del Perú (Hammons 1994), aunque la domesticación del maní cultivado haya sido realizada por indígenas de las tierras bajas tropicales de Sudamérica (Krapovickas 1995).
Antonio Krapovickas (1995) sugiere el origen de la subespecie hypogaea en el Sureste de Bolivia y que la subespecie fastigiata se haya diferenciado más al norte, posiblemente en Perú, donde presenta su mayor variabilidad, con la presencia de las variedades fastigiata, peruviana y aequatoriana (Williams 1989), no descartando la posibilidad de la participación de alguna otra especie silvestre.
Estudios previos en distintas variedades de maní analizadas con técnicas bioquímicas y moleculares tales como isoenzimas, RFLP, RAPD, SSR; no habían reportado variación significativa a nivel del DNA en los genotipos analizados. Sin embargo en los últimos años, se ha logrado encontrar polimorfismos significativos con la ayuda de técnicas como AFLP y recientemente SSR, lo que está permitiendo estructurar un mapa genético en la especie para su uso en el mejoramiento genético del cultivo.
Es necesario que toda especie vegetal posea, unida a ella, información concerniente a su ubicación en determinado tiempo y espacio, para facilitar el proceso de conservación. Esta información geo-referenciada puede ser complementada con otros tipos de datos como clima, suelo, topografía, actividades humanas y demás aspectos del ambiente físico y biológico; con el fin de establecer patrones geográficos de distribución de la diversidad, identificando incluso áreas de alta diversidad, predecir posibilidades de encontrar una especie en áreas que no hayan sido exploradas, así como en la selección y diseño de lugares para la conservación in situ (Guarino et al. 1999).
El Instituto Nacional de Innovación Agraria (INIA) mediante el proyecto: "Modelos de diversidad y de erosión genética en cultivos tradicionales de Perú: asesoría rápida y detección temprana de riesgos usando las herramientas de GIS" recolectó 65 entradas de maní Arachis hypogaea en las cuencas de los ríos: San Alejandro, Ucayali y Aguaytía en la Región Ucayali, las cuales muestran considerable variación fenotípica para importantes características morfológicas como color de grano, forma y reticulado de la vaina, entre otras. (Mori & Mori 2010). Dichas entradas corresponden a 21 variedades locales cultivadas por las comunidades nativas y las colonizadoras.
Se utilizó la técnica AFLP para generar marcadores moleculares y determinar la variabilidad genética de estas 21 variedades locales de maní, en base a sus relaciones de similitud y distancia genética. Asimismo, a partir de los datos de pasaporte y los marcadores moleculares, determinar las áreas geográficas con la mayor riqueza de grupos genéticos haciendo uso del programa de cómputo DIVA.
Materiales y métodoss
Material biológico.-
Se analizaron 65 entradas de maní A. hypogaea (30 entradas pertenecientes a la especie fastigiata y 35 a hypogaea), correspondientes a 21 cultivares nativos del Banco Nacional de Germoplasma de Maní del Instituto Nacional de Investigación y Extensión Agraria del Perú (Tabla 1). Se analizó sólo 1 planta por entrada por ser el maní una especie de reproducción autógama. Dichas entradas sólo corresponden a las colectadas en la región Ucayali dentro de las cuencas mencionadas.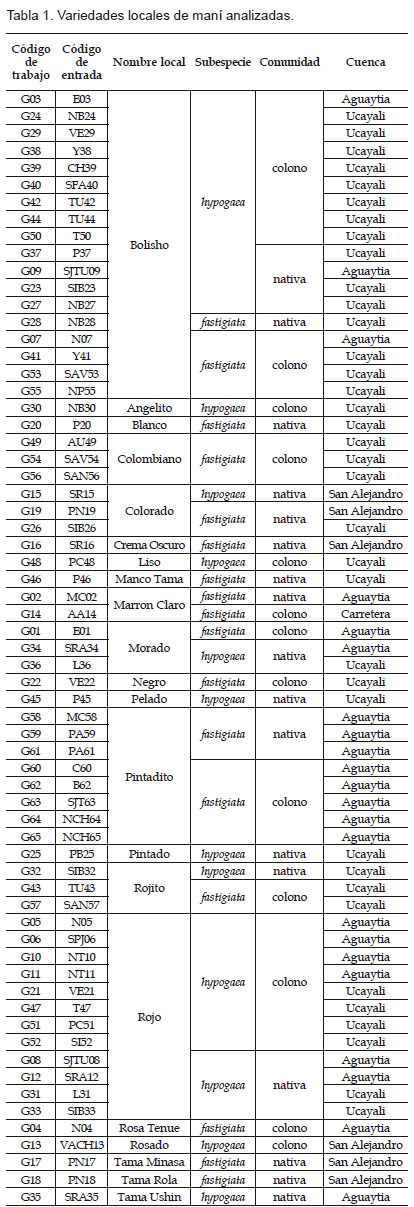
Obtención de marcadores moleculares AFLP.- Para realizar el análisis AFLP se utilizó el kit: AFLP® Analysis System I (Life Technologies 2001), en base a 10 combinaciones de iniciadores (Tabla 2) que mostraron polimorfismo en estudios previos (He & Prakash 1997), los cuales fueron repetidos 2 veces y aplicados según lo indicado en el manual de instrucción (Life Technologies 2001) con algunas modificaciones menores que se detallan a continuación.
Extracción de ADN.- La extracción se realizó mediante el método CTAB (Doyle & Doyle 1990; CIP 1987). Para ello se incrementó la concentración del PVP (polivinilpirrolidona), en el buffer de lisis, del 1 al 2%. El ADN se obtuvo a partir de foliolos sanos de plántulas de maní sembradas en el invernadero.
Cuantificación del ADN.- La cuantificación del ADN se llevó a cabo por comparación, utilizando como patrón de referencia cuantitativa al fago Lambda cortado con la enzima de restricción Pst I, en geles de agarosa al 1% (bromuro de etidio (10mg/mL)), (CIP 1987). El DNA fué posteriormente diluído con buffer TE hasta obtener una concentración aproximada de 25 ng /μL.
Preparación del ADN genómico molde.- Un total de 125 ng de DNA fueron digeridos con 1U de las enzimas de restricción Eco RI y Mse I, durante 2 horas a 37 °C; luego se ligaron los adaptadores a los fragmentos generados en la digestión, durante 2 horas a 20 °C, con lo cual se obtuvo el DNA molde necesario para la preamplificación.
Pre-Amplificación.- Se procedió a realizar una dilución 1/10 (v/v) de la mezcla obtenida anteriormente, con el buffer TE proporcionado en el kit. La dilución obtenida (DNA molde diluido) fue utilizada para realizar la reacción de Pre-Amplificación en el termociclador Perkin Elmer modelo 2400, de acuerdo al programa de amplificación siguiente: 94 °C (30 segundos); 56 °C (60 segundos); 72 °C (60 segundos) durante 20 ciclos. La mezcla resultante conformará el DNA molde pre-amplificado, básico para las amplificaciones con los nucleótidos selectivos.
Amplificación Selectiva.- El DNA molde preamplificado, es diluido en 1/50 (v/v) con el buffer TE. La dilución obtenida es utilizada para las reacción de amplificación selectiva en placas PCR, de acuerdo al programa de amplificación selectiva: Denaturación a 94 °C (60 s), Anillamiento a 65 °C (60 s) y Extensión a 72 °C (90 s) durante un ciclo; luego del cual se reduce la temperatura de Anillamiento en 1 °C para los posteriores ciclos, hasta llegar a los 56 °C (Fase "touch down"). Posteriormente se continúa con 23 ciclos a 94 °C (30 s), 56 °C (30 s) y 72 °C (60 s).
Electroforesis de los productos de amplificación.- Los productos de amplificación fueron separados en geles denaturantes de poliacrilamida (Acrilamida al 6%-Urea 7M) mediante electroforesis vertical en un sistema de secuenciamiento Bio Rad, modelo Sequi-Gen GT, con solución tampón TBE 0,5 X. Se realizó una precorrida a 1600 voltios por 50 minutos.
Se le adicionó a las muestras un tampón de carga (formamida al 96 %) equivalente al 50% del volumen de la muestra amplificada, para luego denaturarlas a 95 °C por 5 minutos y colocadas rápidamente sobre hielo, con la finalidad de evitar posibles renaturaciones, antes de ser cargadas en el gel. Después de denaturar, se cargaron en el gel 8 μL de las muestras amplificadas. El tiempo de corrida electroforética fue de 5 horas a 1700 voltios.
Revelado de los productos de amplificación.- Para visualizar las bandas amplificadas, se utilizó la tinción con nitrato de plata sugerido por Promega (Corporación Promega 1994), con las siguientes modificaciones: solución fría de ácido acético al 10%; enjuagues con agua destilada fría, solución de nitrato de plata al 0,1%. Enjuagar durante 7 segundos con agua destilada fría. El gel es sumergido y agitado en una solución reveladora fría de carbonato de sodio hasta obtener la intensidad y contraste deseados de las bandas AFLPs.
Registro de las bandas.- Las bandas reveladas en la tinción fueron registradas en una matriz de datos, en una hoja Excel de Windows. Se consideró 1, para la presencia de una banda y 0 para la ausencia de la misma.
Análisis de la variabilidad genética.- La Heterocigosidad (H) es ampliamente usada para medir la diversidad alélica o la informatividad de un marcador genético para un locus con varios alelos y es calculada por la fórmula:
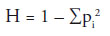
A pesar de ser conceptos diferentes, los términos PIC (Contenido de Información Polimórfica) y H (Heterocigosidad) se utilizan como sinónimos para estimar la "diversidad genética" (Smith et al. 1997; Smith et al. 1992; Raina et al. 2001) y resultaría ser la probabilidad de que un marcador encuentre diferencias entre 2 individuos en al menos 1 locus (Duque 1998). Los valores del PIC varían desde 0 (monomórfico o no discriminatorio) hasta 1 (muy polimórfico o altamente discriminatorio, con varios alelos en igual frecuencia). (Smith et al. 1997).
Sin embargo para un locus con dos alelos se utiliza la siguiente fórmula:
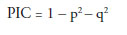
donde "p" es la frecuencia de la presencia del marcador (alelo 1) y "q" es la frecuencia de la ausencia del marcador (alelo 2) (Ghislain et al. 1999; Powell et al. 1996).
Los valores PIC nos dan una idea de la capacidad de cada marcador generado para revelar loci polimórficos en nuestras entradas (Ghislain et al., 1999), así, como el valor de la frecuencia de ocurrencia de cada una de nuestras bandas (alelos) y es considerada además, una medida de diversidad genética (Senior et al. 1998; Smith et al. 1997; Raina et al. 2001).
Obtenidos los valores PIC de cada uno de los marcadores o bandas se procedió con la elección de aquellas cuyo PIC sea >0,1 y <0,5, por ser considerados estos niveles como los límites empíricos para detectar diferencias utilizando un gran número de repeticiones (Ghislain et al. 1999). Las demás bandas fueron descartadas por su bajo contenido de información polimórfica.
Los análisis de asociación en base a las similitudes genéticas se realizaron en el programa NTSYSpc 2.1, utilizando el coeficiente de similitud simple, ya que se asume que cada banda en el gel corresponde a un locus con 2 alelos: presencia (alelo 1) y ausencia de la banda (alelo 2). Al comparar 2 entradas que no posean una determinada banda serán consideradas que poseen el mismo alelo en ese locus (Powell et al. 1996; Ghislain 1999). Sin embargo, otras matrices de similitud fueron generadas en base a los coeficientes de Jaccard y el de DICE, las cuales fueron comparadas con la matriz de similitud simple matching, para encontrar posibles diferencias entre los valores de similitud genética y disminuir el sesgo por la elección del coeficiente de asociación.
Para construir nuestra matriz de similitud se consideró el criterio de selección para marcadores dominantes en base al PIC, el cual sólo considera a aquellos marcadores con frecuencias PIC > 0,10 y < 0,50, con lo cual se redujo el número de marcadores de nuestra matriz de datos, de 157 bandas polimórficas a 135 bandas polimórficas. Esta nueva matriz de datos de 135 bandas polimórficas y 65 entradas fue utilizada para obtener la matriz de similitud genética.
Luego de comparar de par en par todas las entradas, para generar la matriz de similitud, se procedió al análisis de agrupamiento para la obtención del dendrograma y los respectivos clusters. Los análisis de agrupamiento o clusters fueron realizados en base a la matriz de similitud "simple matching" (SM). Se utilizaron tres tipos de ligamiento: simple, completo y promedio, para construir los dendrogramas y visualizar las relaciones de similitud existente en las entradas. Las diferencias entre estos dendrogramas fueron evaluadas mediante la correlación entre los valores cofenéticos de cada dendrograma y la matriz de similitud mediante el test de Mantel.
Para validar el número de grupos generados (clusters) en el dendrograma de acuerdo a cada tipo de ligamiento, se utilizaron 3 controles: correlación entre la matriz de similitud y la matriz cofenética generada en base al dendrograma mediante el test de Mantel; reproducibilidad de los grupos por el análisis "bootstrap", mediante el programa Winboot (Yap 1996), aplicando 500 repeticiones en base al agrupamiento UPGMA; y la estructura de los grupos conformados, el cual nos permitió conocer la proporción de similitud entre los integrantes de un grupo, en base a las bandas compartidas y las bandas exclusivas de dicho grupo (Tabla 3).
Utilizando el programa DIVA-GIS se analizó la distribución de los grupos generados (clusters) en base a las similitudes genéticas, con el objeto de dilucidar patrones geográficos, genéticos, mapeo de riqueza y diversidad basada en datos de marcadores moleculares. Para ello fue necesario utilizar la información registrada en el pasaporte de cada una de las entradas en cuanto a su ubicación geográfica (Latitud, longitud y la altitud).
Se realizó la búsqueda de áreas geográficas que posean la mayor concentración de grupos genéticos (clusters), utilizando para ello celdas (grids) de 0,15° (aproximadamente 225 Km²). Cada grid o cuadrícula representa un área en el cual están ubicadas las entradas colectadas que pertenecen a un determinado grupo genético.
Al comparar la cantidad de grupos genéticos existentes entre una cuadrícula y otra, podemos obtener diferencias entre la variabilidad presente en ellas; así como, ubicar las cuadrículas con el mayor número de grupos genéticos. Cada uno de los grupos obtenidos fueron graficados en sus respectivas zonas geográficas de colecta para observar su distribución.
Resultados
Registro de las bandas informativas amplificadas.- Los patrones de bandas de las combinaciones de "primers" AFLP, de cada una de las entradas, fueron registrados en una matriz de datos en una hoja Excel de Windows. Se consideró 1, para la presencia de una banda y 0 para la ausencia de la misma.
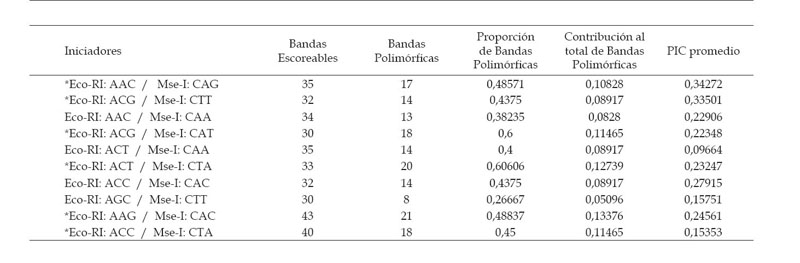
Las 10 combinaciones de iniciadores AFLP amplificaron un total de 344 bandas reproducibles en nuestras 65 entradas, con un promedio de 34 bandas escoreables por combinación de iniciadores (Rango: 30 a 43 bandas) cuyos tamaños se encuentran comprendidas entre los 1100 y 250 pb.
De las 344 bandas reproducibles, sólo 157 bandas fueron informativas o polimórficas (45,6%), que son aquellas que muestran por lo menos 2 estados (alelos): presencia y ausencia del marcador. Las demás bandas son consideradas no informativas o monomórficas y son descartadas del análisis, porque no presentan 2 alelos o estados, por lo tanto no permiten encontrar diferencias entre 2 entradas. Se obtuvo en promedio 16 bandas informativas por combinación de iniciadores (Rango: 08 a 21 bandas).
Contenido del índice polimórfico (PIC).- Los marcadores con valores PIC menores a 0,1 y mayores a 0,5 no fueron considerados, por lo que se eliminaron un total de 22 bandas (14%), las cuales no alcanzaron los valores establecidos. Un total de 135 bandas polimórficas (86%) lograron los niveles de polimorfismo requerido. De estas 135 bandas el 68,8% son generadas por sólo 6 combinaciones de iniciadores, señaladas con asterisco (*) en la Tabla 2.
La combinación de iniciadores Eco-RI: AAC / Mse-I: CAG generó los mayores valores PIC entre nuestras entradas y revela la mejor calidad de bandas informativas. Por otro lado, la combinación de iniciadores Eco-RI: ACT / Mse-I: CAA produce los más bajos valores PIC y una baja calidad en el revelado de los productos de amplificación, al igual que las combinaciones Eco-RI: AAC / Mse-I: CAA y Eco-RI: AGC / Mse-I: CTT.
Los valores PIC promedio de cada combinación de iniciadores fluctúan entre los 0,097 y los 0,343, con un promedio global de 0,23, lo cual indica una variabilidad media, teniendo en consideración que el valor máximo que podría adquirir este índice es de 0,5 para marcadores de tipo dominante.
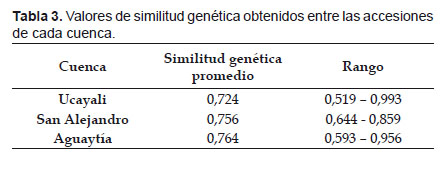
Similitud Genética.- El 47,5% de la población analizada posee una similitud genética comprendida entre los 0,7 y 0,8; el 24,3% de la población posee los mayores valores de similaridad genética, comprendido entre los 0,8 y 1; y el 28,2% de la población posee los menores valores de similitud genética comprendidos entre 0,5 y 0,7, lo cual indica nuevamente una variabilidad media. La mayor variabilidad genética estaría dentro de la cuenca del Ucayali, porque posee los menores valores de similitud genética (Tabla 3); sin embargo, dichos valores son muy cercanos al de las otras cuencas.
Ligamiento completo.- El dendrograma de ligamiento completo estableció la conformación de 8 grupos genéticos a un nivel de similitud de 0,65 (Fig. 1). Se puede notar cierta tendencia en las accesiones de agruparse de acuerdo al tipo de suelo en la cual fueron colectadas, ya que el 85% de las entradas colectadas en las zonas denominadas de "altura" (162 – 367 m de altitud) están ubicadas en el cluster 8, el cual es el más numeroso. El 63,2% de las entradas fueron colectadas en terrenos "bajos" (132 – 152 m), sin embargo, la mayoría de ellas pertenecen a diferentes clusters.
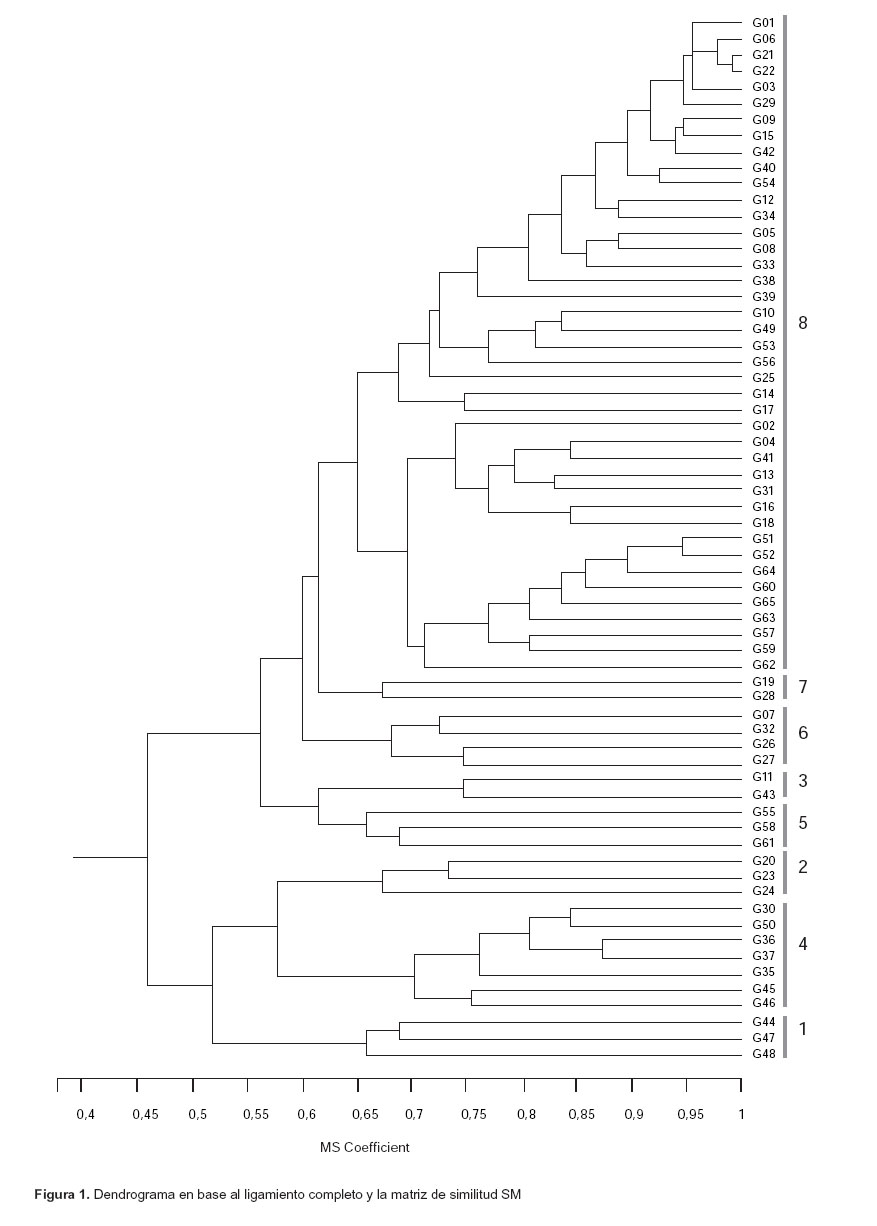
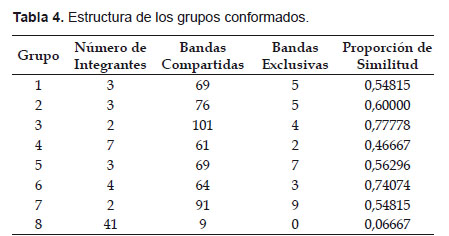
El 87,5% de los grupos están fuertemente estructurados, al ser similares entre sí los miembros de un grupo en un rango del 47 al 76%; excepto el grupo 8, el cual sólo alcanza un 7% de bandas compartidas (Tabla 4). Es interesante hacer notar que el 54% de los miembros de este grupo 8 están ubicados en zonas denominados de "altura" entre los 162 y 367 msnm, al igual que los grupos 3 y 5 hallados entre los 175 y 303 m de altitud.
Distribución espacial de la variabilidad genética del maní.- Las entradas de maní fueron colectadas en 7 distritos pertenecientes a 2 provincias (Coronel Portillo y Padre Abad) abarcando 3 cuencas y a 4 tipos distintos de suelos. La mayor cantidad de colectas (52%) se registran en el distrito de Callería, provincia Coronel Portillo, cuenca del Ucayali, cuyos suelos son predominantemente "bajos" (Barrizal, Playa y Restinga).
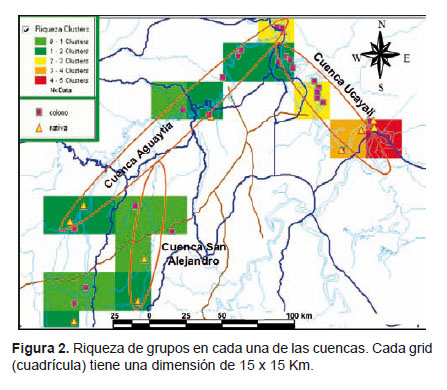
Riqueza de grupos.- La mayor riqueza de grupos genéticos de maní se encuentran en la cuenca del Ucayali (Fig. 2), distrito de Callería, provincia Coronel Portillo del departamento de Ucayali. En dicha cuenca se logra ubicar los 8 grupos genéticos, siendo los grupos 1 y 3 exclusivos de esta cuenca, con una riqueza de hasta 5 grupos diferentes en una solo celda. En la cuenca del Aguaytía se logra ubicar hasta 5 grupos genéticos y en la cuenca del San Alejandro sólo se tiene 2 grupos. Es importante mencionar que todas las entradas de la Cuenca San Alejandro pertenecen al grupo 8.
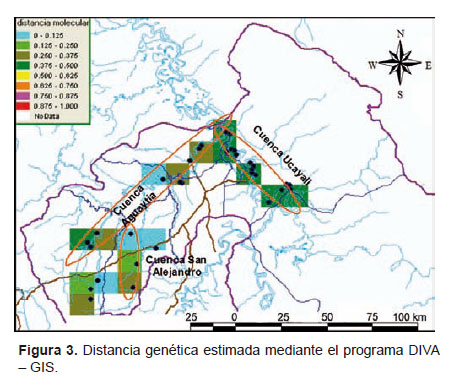
Distancia genética.- Los datos de marcadores moleculares, asociados a la ubicación geográfica de las localidades, fueron analizados también con el programa DIVA-GIS con la finalidad de estimar la variabilidad genética entre nuestras entradas. Para ello se calcularon las distancias genéticas en base al coeficiente de Sokal & Michener (1958).
Los valores de distancia genética se encuentran comprendidos entre 0> y <0,5, lo cual indica una variabilidad predominantemente media; siendo la cuenca del Ucayali la que posee los mayores valores de distancia genética (0,375 – 0,500). (Fig. 3).
Discusión
Variabilidad genética.- A pesar de que el maní posee una gran variabilidad morfológica (Kochert et. al. 1996; Williams 1989), dicha variabilidad no había podido ser demostrada molecularmente al utilizar diversas técnicas de marcadores para encontrar polimorfismo a nivel del ADN en el maní (Garcia et al. 1 995; Halward et al. 1991; Halward et al. 1992; Hopkins et al. 1999; Kochert et al. 1991; Subramanian et al. 2000).
Sin embargo algunos investigadores lograron encontrar polimorfismo genético a nivel del ADN del maní, empleando la técnica de marcadores AFLP (He & Prakash 1997), con lo cual fue posible estimar las relaciones genéticas entre las diferentes variedades botánicas del cultivo (He & Prakash, 2001), aunque anteriormente, habían sido estimadas las relaciones entre algunas especies silvestres del mismo (He et al. 1995).
Al trabajar nosotros con las 10 combinaciones de iniciadores AFLP que mostraron ser polimórficos en estudios previos, obtuvimos algunas diferencias. Las 10 combinaciones de iniciadores amplificaron un total de 344 bandas, de las cuales 157 fueron informativas; lo cual difiere de las 695 bandas totales y 53 informativas reportadas anteriormente. (He & Prakash 1997).
Dicha diferencia podría ser explicada por el número de entradas de maní evaluadas, ya que en dicho estudio sólo se trabajo con 6 entradas de maní, pertenecientes a 3 variedades botánicas, en cambio nosotros trabajamos con 65 entradas de maní, lo que tal vez haya facilitado a la obtención de un mayor número de bandas informativas.
Los valores PIC nos dan una medida de la variabilidad genética (Powell et. al. 1996; Agrama & Tuinstra, 2003), pero por ser los marcadores AFLP dominantes, no muestran la totalidad de alelos que existen en un locus, por lo que podríamos obtener resultados diferentes si analizamos nuestras muestras con marcadores codominantes. Sin embargo el gran poder de análisis multiloci de los marcadores AFLP nos permite encontrar un mayor número de diferencias y similitudes a nivel del ADN, con lo cual es posible obtener grupos genéticamente similares al aplicar técnicas de taxonomía numérica en el análisis de nuestros resultados.
Los valores PIC fueron de gran utilidad para la elección de las bandas informativas tomadas en cuenta para el análisis de similitud genética, porque nos permitió establecer las bandas que detectan diferencias entre nuestras entradas, para establecer un matriz de similitud genética. (Ghislain et al. 1999).
El coeficiente de asociación simple considera las presencias y ausencias de un marcador como similitudes genéticas. (Powell et al. 1996; Ghislain et al. 1999); sin embargo la ausencia de un marcador no implica necesariamente una similitud genética, por lo tanto algunos investigadores utilizan los coeficientes de Jaccard y DICE, los cuales sólo consideran las presencias de las bandas como una similitud genética. (Vipa & Huestis 1997; Erschadi et. al. 2 000; Teulat et. al. 2000).
Por ello, se generaron matrices de similitud genética utilizando diferentes coeficientes de asociación para establecer si existían diferencias entre las matrices de similitud generadas. Los altos niveles de correlación entre las distintas matrices nos permitió determinar que no existían mayores diferencias entre ellas, por lo tanto podríamos trabajar indistintamente con cualquier tipo de coeficiente de asociación genética, sin que ello ocasione grandes distorsiones al realizar nuestro análisis de agrupamiento o dendrograma.
Los análisis de agrupamiento son realizados principalmente con el ligamiento promedio, sin embargo nosotros optamos por el ligamiento completo (a pesar de que su correlación cofenética no es buena), porque fue el único dendrograma que nos permitió distinguir los 8 grupos genéticos o clusters generados en el análisis de reproducibilidad "bootstrap". Además los dendrogramas generados por los ligamientos simple y promedio no establecieron una clara distinción de grupos entre las entradas, revelados en el análisis "bootstrap".
Distribución espacial de la variabilidad genética.- El programa DIVA – GIS, permitió visualizar la distribución espacial de los grupos genéticos obtenidos, los cuales están distribuidos principalmente en la cuenca del Ucayali. Al parecer dicha distribución obedece principalmente al tipo de suelo (barrizales y playas) de la cuenca del Ucayali, el cual es inundable durante la temporada de crecida de los ríos, lo que permite sembrar en un terreno suave, ideal para el desarrollo de las vainas del maní, posibilitando un mayor número de comunidades dedicadas a su cultivo; a comparación de las comunidades de las zonas de "altura", la cuales no son inundadas por los ríos, por lo que el terreno es duro y compacto, limitando el desarrollo de las vainas. (David Williams, comunicación personal).
Sin embargo, en la cuenca del Ucayali se efectuó la mayor cantidad de colectas (52 %) lo que tal vez haya influido en que la zona agrupe a la mayor cantidad de grupos, pero no debemos dejar de considerar que 2 grupos, son exclusivos de la cuenca. Los valores de distancia genética generados por el programa DIVA – GIS, reafirma a la cuenca del Ucayali como la que posee la mayor variabilidad genética del cultivo de maní.
Literatura citada
Agrama H. A. & M. R. Tuinstra. 2003. Phylogenetic diversity and relationships among sorghum accessions using SSRs and RAPDs. African Journal of Biotechnology 2 (10): 334-340. [ Links ]
CIP, Centro Internacional de la Papa. 1987. Protocolos de laboratorio de biología molecular-tipificación genética. Ghislain M., D. Zhang y M. Herrera. Ed. Departamento de Recursos Genéticos. Manual de capacitación CIP. Lima Perú. [ Links ]
Corporación Promega. 1994. Staining Nucleic Acids with Silver: An alternative to radioisotopic and fluorescent labeling. Promega notes magazine. 45: 13-19. [ Links ]
Doyle J. & J. Doyle. 1990. Isolation of Plant DNA from fresh tissue. Focus 12:13-15. [ Links ]
Duque M. C. 1998. Introducción al analisis de datos moleculares. Manual de capacitación. Edit. CIAT. Cali – Colombia. [ Links ]
Erschadi S., G. Haberer, M. Schöniger & R. Torres-Ruiz. 2000. Estimating genetic diversity of Arabidopsis thaliana ecotypes with amplified fragment length polymorphism (AFLP). Theoretical and Applied Genetics 100: 633-640. [ Links ]
Garcia G., H. Stalker & G. Kochert. 1995. Introgression analysis of an interspecific hybrid population in peanuts (Arachis hypogaea L.) using RFLP and RAPD markers. Genome 38: 166-176. [ Links ]
Ghislain M., D. Zhang, D. Fajardo, Z. Huaman & R. Hijmans. 1999. Marker-assisted sampling of the cultivated Andean potato Solanum phureja collection using RAPD markers. Genetic Resources and Crop Evolution 46: 547-555. Holanda. [ Links ]
Guarino L., N. Maxted & M. Sawkins.1999. Analysis of geo-referenced data and the conservation and use of plant genetic resources. S.L. Greene & L. Guarino (editors). Linking genetic resources and geography: emerging strategies for conserving and using crop biodiversity. American Society for Agronomy Special Publication 27. ASA, CSSA, and SSSA, Madison, Wisconsin. [ Links ]
Halward T., M. Stalker, E. La Rue & G. Kochert. 1991. Genetic variation detectable with molecular markers among unadapted germplasm resources of cultivated peanut and related wild species. Genome, 34:1013-1020. Canada. [ Links ]
Halward T., M. Stalker, E. La Rue & G. Kochert. 1992. Use of single-primer DNA amplifications in genetics studies of peanut (Arachis hypogarea L.). Plant Molecular Biology 18:315-325. [ Links ]
Hammons R. O. 1973. Genetics of Arachis hypogaea. Peanuts: culture and uses. American Peanut Research and Education Association, 135-173. [ Links ]
Hammons R. O. 1994. The origin and early history of the peanut. p. 24–42. In J. Smartt (ed.) The peanut crop: A scientific basis for improvement. Chapman and Hall, London – Inglaterra. [ Links ]
He G. & C. Prakash. 1997. Identification of polymorphic DNA markers in cultivated peanut (Arachis hypogaea. L). Euphytica 97: 143-149. Holanda. [ Links ]
He G. & C. Prakash. 2001. Evaluation of genetic relationships among botanical varieties of cultivated peanut (Arachis hypogaea L.) using AFLP markers. Genetic Resources and Crop Evolution 48: 347-342. Holanda. [ Links ]
He G., K. Singh & C. Prakash. 1995. Análisis de las relaciones genéticas entre accesiones de A. stenosperma y A. duranensis , usando marcadores DNA. Center for Plant Biotechnology Research. University Tuskeege. ICRISAT. [ Links ]
Hopkins M., A. Casa, T. Wang, S. Mitchell, R. Dean, G. Kochert y S. Kresovich. 1999. "Discovery and Characterization of Polimorphic Simple Sequence Repeats (SSRs) in Peanut". Crop Science. 39: 1243-1247. [ Links ]
Kochert G., T. Halward y T. Stalker. 1996. Genetic variation in peanut and its implications in plant breeding. B. Pickersgill and J. M. Lock editors. Advances in Legume Systematics 8: Legumes of economic importance. pp. 19-30. Royal Botanical Gardens. Kew. [ Links ]
Kochert G., T. Halward, W. Branch & C. Simpson .1991. RFLP variability in peanut Arachis hypogaea L.) cultivars and wild species. Theoretical and Applied Genetics. 81: 565-570. [ Links ]
Krapovickas A. 1995. El origen y dispersión de las variedades del maní. Academia Nacional de Agronomía y Veterinaria. Tomo XLIX: 18-26. Corrientes - Argentina. [ Links ]
Life Technologies. 2001. Manual de Instrucción. AFLP® Analysis System I, AFLP Starter Primer Kit. GIBCO. [ Links ]
Mori J.A. & y Mori C. 2010. Análisis de la Variabilidad Genética del maní culivado (Arachis hypogaea) del distrito de Iparia, Rio Ucayali . Peru. Proceedings Primer Congreso de Mejoramiento gemnético y Biotecnología Agrícola, pp 17-19. [ Links ]
Powell W., M. Morgante, C. Andre, et al. 1996. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Molecular Breeding 2: 225-238. [ Links ]
Raina S., V. Rani, T. Kojima, Y. Ogihara, K. Singh & R. Devarumath. 2001. RAPD and ISSR fingerprintings as useful genetic markers for analysis of genetic diversity, varietal identification and phylogenetic relationships in peanut (Arachis hypogaea L.) cultivars and wild species. Genome 44: 763-772. [ Links ]
Sauer J.D. 1993. Historical geography of crop plants. Edit. CRC Press Boca Raton, Florida – Estados Unidos. [ Links ]
Senior M. L., J. P. Murphy, M. M. Goodman & C. W. Stuber. 1998. Utility of SSRs for determining genetic similarities and relationships in maize using an agarose gel system. Crop Science. 38: 1 088-1 098. [ Links ]
Singh A. & J. P. Moss. 1982. Utilization of wild relatives in genetic improvement of Arachis hypogaea. Parte 2. Chromosome complements of species in section Arachis. Theoretical and Applied Genetics 61: 305-314. [ Links ]
Smith O., J. Smith, S. Bowen & R. Tenborg. 1992. Number of RFLP probes necessary to show associations between lines. Maize Genet. Coop. Newsletter. 66:66 [ Links ]
Smith, J., E. Chin, Ll. Shu, O. Smith, S. Wall, M. Senior, S. Mitchell, S. Kresovich & J. Ziegle. 1997. An evaluation of the utility of SSR loci as molecular markers in maize (Zea mays L.): comparisons with data from RFLPs and pedigree. Theoretical and Applied Genetics. 95: 163-173. [ Links ]
Sokai R.R. & Michener C.D. 1958. A statistical method for evaluating systematic relationships. Univ Kansas Sci. Bull 38:1409 -1438. [ Links ]
Stalker H. & C. Chapman. 1989. Management of Germplasm: Characterization, Evaluation and Enhancement. IBPGR. Training Courses. Lecture Series 2. Italia. [ Links ]
Subramanian V., S. Gurtu, R. Nageswara & S. Nigam. 2000. Identification of DNA polymorphism in cultivated groundnut using random amplified polymorphic DNA (RAPD) assay. Genome 43: 656-660. Canada. [ Links ]
Teulat B., C. Aldam, R. Trehin, P. Lebrun, J. Barker, G. Arnold, A. Karp, L. Baudouin & F. Rognon. 2000. An analysis of genetic diversity in coconut (Cocos nucifera) populations from across the geographic range using sequence-tagged microsatellites (SSRs) and AFLPs. Theoretical and Applied Genetics 100: 764-771. [ Links ]
Vipa H. & G. Huestis. 1997. Amplified fragment polymorphismas a tool for DNA fingerprinting sunflower germplasm: genetic diversity among oilseed inbred lines. Theoretical and Applied Genetics 95: 400-407. [ Links ]
Williams D. E. 1989. Exploration of Amazonian Bolivia yields rare peanuts landrace. Diversity 5(4): 12-13. [ Links ]
Yap I. V. & R. J. Nelson. 1996. Winboot: A program for performing bootstrap analysis of binary data to determine the confidence limits of UPGMA-based dendrograms. IRRI Discuss. Pap. Ser. 14. [ Links ]
Email: Luis Rimachi: lrimachi@inia.gob.pe
Rolando Estrada: restradaj@gmail.com
Presentado: 24/07/2012
Aceptado: 29/11/2012
Publicado online: 15/01/2013
