










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkRevista Peruana de Biología
versión On-line ISSN 1727-9933
Rev. peru biol. vol.20 no.3 Lima dic. 2013
TRABAJOS ORIGINALES
Identificación de genes relacionados a sequía en papas nativas empleando RNA-Seq
Identification of genes related to drought in native potatoes using RNA-Seq
Yerisf Torres*, Roberto Lozano, Carlos Merino y Gisella Orjeda
Unidad de genómica, Laboratorios de Investigación y Desarrollo (LID), Universidad Peruana Cayetano Heredia. Av. Honorio Delgado 430, Urb. Ingeniería, S.M.P. Lima - Perú.
Email Yerisf Torres: yerisf.torres.a@upch.pe
Email Roberto Lozano: roberto.lozano@upch.pe
Email Carlos Merino: carlos.merino.m@upch.pe
Email Gisella Orjeda: gisella.orjeda@upch.pe
Resumen
El reciente desarrollo del RNA-Seq, un método de secuenciamiento masivo en paralelo para el análisis de transcriptomas permite conocer el perfil de expresión de las plantas en respuesta a estrés de tipo abiótico y biótico. En este estudio, se secuenció el mRNA proveniente de hojas y raíces de dos variedades de papas nativas expuestas a diferentes niveles de sequía. Lecturas o reads de 50 pares de bases provenientes de mRNA se mapearon al genoma de papa: 75 – 82% mapearon a posiciones únicas, 6 – 14% mapearon a múltiples posiciones y 9 – 12% no mapearon a posición alguna del genoma. Comparando los perfiles de expresión, se encontraron entre 887 – 1925 genes inducidos/reprimidos por sequía en la variedad sensible y 998 – 1995 en la tolerante. Este estudio generó información de gran valor que podrá ser utilizada en futuros estudios para comprender mejor los mecanismos moleculares de resistencia a sequía en papa y especies cercanas.
Palabras clave : RNA-Seq; transcriptoma; sequía; papa; mRNA.
Abstract
The recent advent RNA sequencing technology (RNA-Seq), a massively parallel sequencing method for transcriptome analysis, provides an opportunity to understand the expression profile of plants in response to biotic and abiotic stress. In this study, the mRNA was sequencing from leaves and roots of two native potato varieties at different levels of drought. Fifty-base-pair reads from whole mRNAs were mapped to the potato genomic sequence: 75 – 82% mapped uniquely to the genome, 6 – 14% mapped to several locations in the genome and 9 – 12% had no match in the genome. Comparing expression profiles, 887 to 1925 genes were found to be induced/repressed by drought in the sensible variety and 998 to 1995 in the tolerant. This research provides valuable information for future studies and deeper understanding of the molecular mechanism of drought resistance in potato and related species.
Keywords: RNA-Seq; transcriptome; drought; potato; mRNA.
Introducción
La sequía es el estrés medio ambiental más importante en la agricultura, por lo que se vienen realizado muchos esfuerzos para mejorar la productividad de los cultivos bajo condiciones limitantes de agua. La papa, el tercer cultivo alimentario más importante del mundo (FAOSTAT 2008) es muy sensible a la sequía, ya que necesita un riego frecuente. A diferencia de Solanum tuberosum subs. tuberosum, las especies nativas de los andes, cultivadas a altitudes de 3500 m, están adaptadas a diversas condiciones climáticas adversas (Vasquez-Robinet et al., 2008). Esto hace a estas variedades de papa los candidatos ideales para estudiar la expresión de genes responsables de la tolerancia a la sequía.
Este trabajo pretende identificar genes relacionados a sequía en papa, empleando la tecnología de secuenciamiento de RNA (RNA-Seq), una herramienta revolucionaria de la transcriptómica (Mortazavi et al. 2008) que permite secuenciar genes transcritos y cuantificarlos de manera precisa.
Materiales y métodos
Material vegetal .- Dos variedades de S. tuberosum andigena, Negrita 703671 (tolerante a sequía) y Wila-HuakaLajra 703248 (susceptible a sequía) fueron propagadas in-vitro, enraizadas y transferidas a un sistema aeropónico de cultivo en la Estación Experimental de INIA- Huancayo.
Diseño del experimento.- Las plantas recibieron una irrigación normal durante aproximadamente 3 meses, tiempo después del cual comienza la tuberización, en este momento se inició la inducción del estrés por sequía dejando de regar a las plantas.
Se tomaron muestras de hojas y raíces de ambas variedades considerando cuatro tiempos de muestreo. Para determinar los tiempos de muestreo se realizaron mediciones de la tasa fotosintéticas de la variedad tolerante (703671) en condiciones normales y de sequía, empleando el equipo CI-340 Handheld Photosynthesis System (CID Bio-Science, Inc. USA) que es un analizador infrarrojo de CO2/H2O que mide la cantidad de CO2 asimilada por un área de hoja conocida en un tiempo dado. El Tiempo Cero correspondió al control del experimento, antes del inicio de la sequía. Los tiempos T1, T2 y T3 correspondieron a una disminución de la tasa fotosintética en la planta tolerante de 25%, 50 – 60% y una recuperación hasta del 80% del valor control respectivamente (Vásquez- Robinet et al. 2008). Los tiempos T1, T2 representaron la respuesta temprana y tardía de las plantas frente a la sequía, mientras que el tiempo T3 represento la recuperación de las plantas luego de reiniciado el riego.
Análisis de expresión.- El RNA fue aislado de aproximadamente 1 – 2 g de tejido de hojas y raíces usando el método fenol-cloroformo (Buell Lab - Michigan State University, comunicación personal 2010), y purificado usando el Kit Ambion DNA-free (Cat. No. 1906).
El RNA extraído fue secuenciado empleando un secuenciador Illumina Hi-SeqTM 2000. Brevemente consiste en que la población de RNA extraído fue convertido a una librería de fragmentos de DNA, cada molécula luego fue amplificada, cuantificada y secuenciada, con lecturas de 50 pares de bases (bp). Este procedimiento fue realizado en la Universidad de Michigan (Buell Lab, Michigan State University). Se generaron en total 16 librerías correspondientes a mRNA de hojas y raíces de las dos variedades en estudio en los cuatro tiempos de muestreo (Tabla 1).
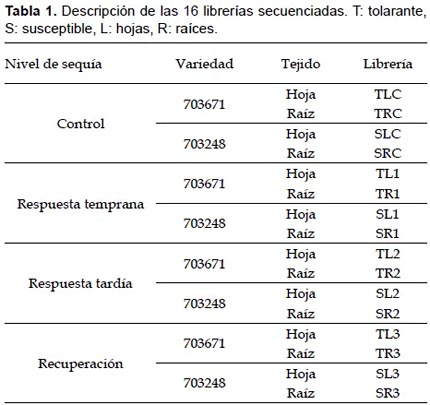
Para el análisis bioinformático se realizó primero un control de calidad de las lecturas empleando el programa FastQC versión 0.10.0, y dos herramientas de FastX, FASTQ Clipper, que eliminan secuencias de adaptadores o linkers y FASTQ Trimmed, que permite eliminar bases de mala calidad presentes en las lecturas, acortando el tamaño de éstas.
Las lecturas fueron alineadas al genoma de referencia de papa (PGSC. 2011), empleando los programas Bowtie 2.0.0 (Langmead et al. 2009) y TopHat (Trapnell et al. 2009). Posteriormente se ensamblaron y cuantificaron las lecturas empleando el programa Cufflinks 1.3.0 (Trapnell et al., 2010), y se empleó la herramienta Cuffdiff para identificar los genes con cambios más significativos en el nivel de expresión durante el experimento.
Resultados y discusión
Control de calidad.- Con el programa FastQ se determinó el Phred Score para los reads de las 32 librerías. Los valores Phred varían entre 4 y 60, y asignaron un valor de calidad a cada base de una secuencia, siendo más alta la calidad al más alto valor. Se consideró como umbral un valor Phred > 28 (considerando que un valor de Phred = 30 indica que la probabilidad de tener una base incorrecta es de 1 en 1000). En 5 librerías las 50 bases de los reads tienes un valor Phred por encima del umbral, 6 librerías presentaron las 3 primeras bases con un valor Phred debajo del umbral y 5 librerías presentaron las 4 primeras bases con un valor Phred debajo del umbral. En estos dos últimos casos se empleó la herramienta FastX-Trimmer para cortar las primeras 3 ó 4 bases de todos los reads de estas librerías y optimizar la calidad de los mismos.
Mapeo de las lecturas.- Una vez realizado el control de calidad, se empleó el programa TopHat para mapear todas las secuencias obtenidas al genoma de referencia de papa que corresponde a S. tuberosum Group Phureja DM1-3 516R44 (PGSC 2011). En base al alineamiento, las secuencias fueron clasificadas en tres clases: reads únicos que son aquellos que mapean con una única posición en el genoma, reads que mapean con más de una posición en el genoma de referencia y los reads que no mapean con región genómica alguna. Empleando el script Perl mapping stadisticas se calculó el número y porcentaje de estos reads (Tabla 2).
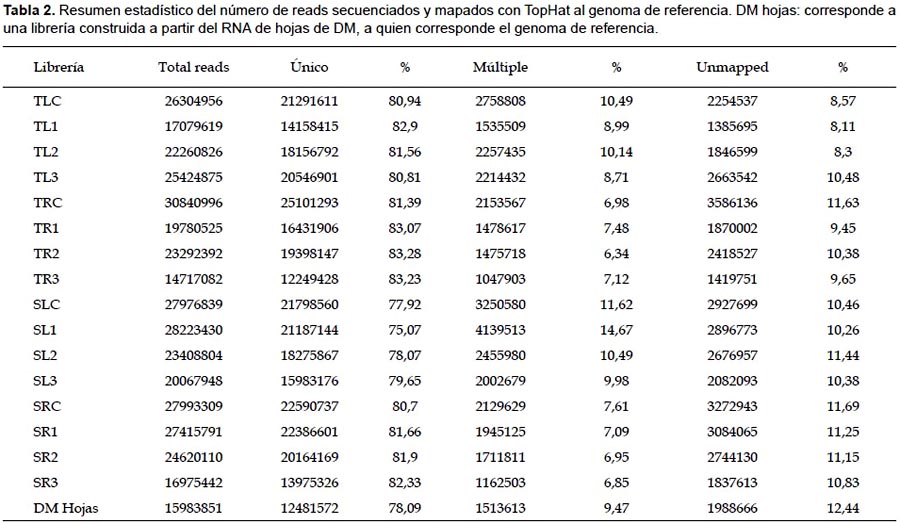
Del total de reads evaluados por cada librería, el 75 – 83% de reads fueron únicos, mientras que el 6 – 14% mapearon a múltiples localizaciones en el genoma y 8 – 11% no mapearon con el genoma. Estos valores indican un desempeño/ rendimiento similar de construcción y secuenciamiento entre las librerías. Adicionalmente, como un control de calidad se secuenció una librería construida a partir de hojas del individuo de referencia DM1-3 516R44, encontrándose valores semejantes.
Expresión diferencial de genes.- El siguiente paso en el análisis de RNA-Seq fue la reconstrucción del transcriptoma y su cuantificación El nivel de expresión de todos los transcriptos que mapearon en el genoma fue cuantificado como fragmentos por kilobase de exón por millón de reads (FPKM).
La comparación de niveles de expresión entre diferentes muestras es parte clave del secuenciamiento de transcriptoma. Empleando el programa Cuffdiff se compararon los perfiles de expresión de las variedades susceptible (703248) y tolerante (703671) durante la exposición a sequía (respuesta temprana, tardía y recuperación) y además entre ambos individuos.
En la Figura 1 se muestra la comparación del número total de transcriptos diferencialmente expresados en hojas y raíces de 703248 y 703671 bajo los tratamientos de sequía; el número de genes diferencialmente expresados entre los tratamientos varía entre 887 – 1995 y se encontró un aumento de este número conforme avanza el experimento.

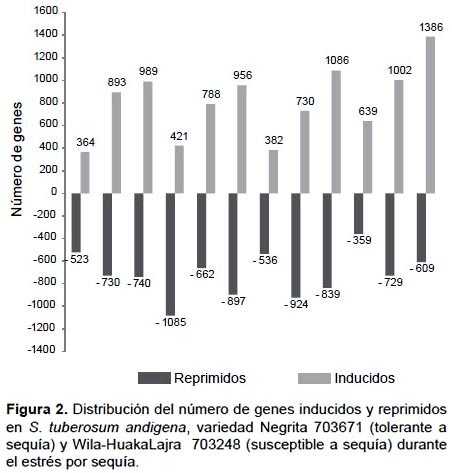
Respecto a la inducción y represión génica, en hojas de 703248 durante la respuesta temprana se encontró que 364 genes fueron inducidos significativamente, mientras que en la sequía tardía y en la recuperación 893 y 989 genes respectivamente; esta tendencia creciente en la inducción de genes se mantiene en hojas y raíces de ambas variedades conforme la sequía avanza. Por otra parte, el número de genes reprimidos varió entre 359 en raíces de 703671 durante la respuesta temprana y 1085 en hojas de 703671 también en la respuesta temprana (Figura 2).
Comparando el perfil de expresión de ambas variedades, se encontró que en la variedad 703671 un mayor número de genes mostraron cambios significativos en respuesta a la sequía. En la respuesta temprana, 149 genes inducidos y 276 reprimidos fueron comunes a hojas de las dos variedades bajo estudio. Se encontró un mayor número de genes específicos y diferencialmente expresados en hojas de la variedad 703671 (272 inducidos y 809 reprimidos), mientras que se encontraron solo 562 genes (215 inducidos y 247 reprimidos) específicos de hojas de la variedad 703248 (Figuras 3 y 4). Un patrón similar se encontró en raíces, 170 genes inducidos y 101 reprimidos fueron comunes en las dos variedades, encontrándose un mayor número de genes específicos y diferencialmente expresados en la variedad 703671 (469 inducidos y 983 reprimidos), mientras que se encontraron solo 646 genes (211 inducidos y 435 reprimidos) específicos de la variedad 703248 (Figs. 3 y 4). Estos genes específicos de la variedad tolerante representan candidatos que deberán ser estudiados a mayor profundidad para conocer el rol que estos cumplen en la resistencia a sequía.
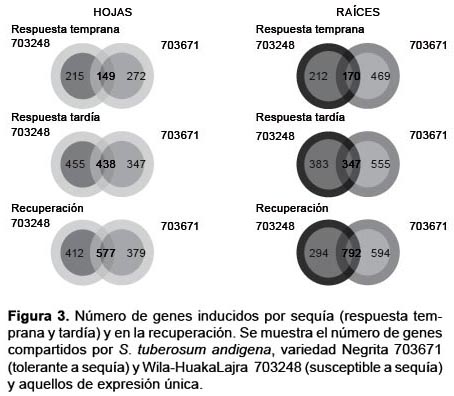
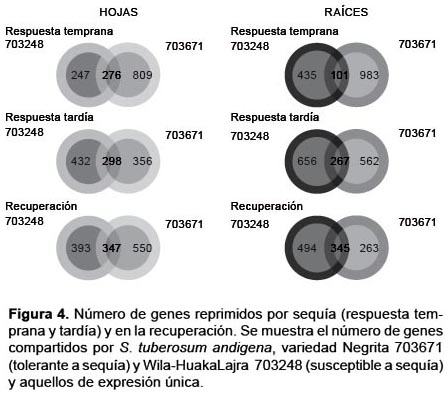
Es importante resaltar que, durante la respuesta temprana de ambas variedades se encontró un mayor número de genes únicos y específicos con expresión diferencial en comparación al de los genes comunes, mientras que en la etapa de recuperación es mayor el número de genes comunes con expresión diferencial. Lo cual podría indicar que los mecanismos de respuesta temprana a sequía son específicos de cada variedad, pudiendo ser esta la etapa clave para encontrar genes responsables de la resistencia a sequía.
En este trabajo, el empleo del RNA-Seq permitió generar 400 millones de reads, que analizados y procesados con herramientas bioinformáticas permitieron identificar y cuantificar un gran número de genes de papa relacionados a sequía y que podrían permitir comprender mejor los mecanismos de resistencia en papa y otras especies relacionadas.
Información adicional
Contribución de autores.- Gisella Orjeda y Yerisf Torres: concepción y diseño de experimento. Yerisf Torres, Roberto Lozano: realización del experimento. Yerisf Torres, Roberto Lozano y Carlos Merino: análisis de datos. Yerisf Torres, Roberto Lozano y Carlos Merino: redacción del artículo.
Conflicto de interés.- Los autores han declarado no incurrir en conflicto de intereses.
Financiamiento.- Los autores agradecen el financiamiento FINCyT (099-FINCyT-EQUIP-2009)/(076-FINCyT-PIN-2008).
Literatura citada
FAOSTAT 2008. Potato world: Production and consumption. International Year of the Potato. <http://www.potato2008.org/en/world/>. Acceso: 08/10/2008. [ Links ]
Langmead B., C. Trapnell, M. Pop & S. Salzberg. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol.10:R25.
Mortazavi A., B. Williams, K. McCue, L. Schaeffer & B. Wold 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods, Vol. 5, Isuue7, pp. 621-628.
PGSC (The Potato Genome Sequencing Consortium). 2011. Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195. [ Links ]
Trapnell C., L. Pachter & SL. Salzberg. 2009. TopHat: discovering splice junctions with RNA-seq. Bioinformatics.;25:1105–1111.
Trapnell C., A. Roberts, L. Goff, et al. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, Vol. 7, Issue 3, pag 562–578.
Vasquez-Robinet C., Sh. Mane, A. Ulanov, et al. 2008. Physiological and molecular adaptations to drought in Andean potato genotypes. Journal of Experimental Botany, Vol. 59, No. 8, pp. 2109–2123.
Presentado: 05/11/2013
Aceptado: 20/01/2014
Publicado online: 14/03/2014
Ciudad Universitaria, Av. Venezuela cda. 34 s/n
Lima 1 - Perú
Casilla Postal 11-0058. Lima 11- Perú
Telf. (0511) 452-4135
Telefax (0511( 464-9110

lromeroc@unmsm.edu.pe