










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkRevista Peruana de Biología
versión On-line ISSN 1727-9933
Rev. peru biol. vol.24 no.2 Lima mayo/agos. 2017
http://dx.doi.org/10.15381/rpb.v24i2.12807
TRABAJOS ORIGINALES
High genetic connectivity and Population Expansion of Scomber japonicus in the Northern Humboldt Current System revealed by mitochondrial control region sequences
Alta conectividad genética y expansión poblacional de Scomber japonicus en en la parte norte del Sistema de Corriente de Humboldt reveladas por secuencias de la región control mitocondrial
Sergio Barahona*, Daniel Oré-Chávez and Roger Quiroz Bazán
Universidad Nacional Mayor de San Marcos, Facultad de Ciencias Biológicas, Laboratorio de Ecología Molecular y Biodiversidad Acuática. Ciudad Universitaria, Av. Venezuela Cdra 34 s/n, Lima 1-Perú.
*Corresponding author
Email Sergio Barahona: srgbarahona89@gmail.com
Email Daniel Ore-Chavez: daniel.ore@unmsm.edu.pe
Email Roger Quiroz Bazan: rquirozb@unmsm.edu.pe
Abstract
The chub mackerel, Scomber japonicus supports an important fishery in the Southeast Pacific, however, its population genetics is currently unknown. In the present study, we examined the genetic structure, the gene flow and the historical demography of this species in the Northern Humboldt Current System. Samples were collected during summer of years 2013-2014 from three fishing points (Paita, Ventanilla and Ilo), covering 12 degrees of latitude along the coast of Peru. A 532 bp segment of the mitochondrial control region was sequenced in 72 individuals, which allowed us to detect a total of 29 polymorphic sites, 35 haplotypes, moderate-to-high levels of haplotype diversity (0.793 – 0.969) and very low levels of nucleotide diversity (0.004 – 0.008). Gene flow analysis showed high levels of connectivity among populations in the sampling areas. Analysis of molecular variance (ФST = 0.00868, P = 0.1837), population pairwise ФST comparisons and genetic differentiation tests confirmed the lack of genetic structuring among the three localities. These analyses suggest that sampling sites analyzed can be considered as a single gene pool. Migratory behavior, the high dispersal potential of early stages and the lack of oceanographic barriers may explain its genetic homogeneity along the Peruvian sea. Historical demography was also examined. Neutrality tests, mismatch distribution and Bayesian skyline plot suggested a population expansion scenario that took place during the Late Pleistocene. This study provides novel information on population genetics of the chub mackerel in the Southeast Pacific.
Keywords: Southeast Pacific; Peruvian sea; fish genetics; gene flow; historical demography.
Resumen
La caballa, Scomber japonicus soporta una pesquería importante en el Pacífico Sudeste, sin embargo, su genética de poblaciones se desconoce actualmente. En el presente estudio se examinó la estructura genética, el flujo génico y la demografía histórica de esta especie en la parte norte del Sistema de la Corriente de Humboldt. Las muestras fueron colectadas en los veranos del 2013 y 2014 en tres puntos de desembarco de pesca (Paita, Ventanilla e Ilo) cubriendo 12 grados de latitud frente a la costa peruana. Se secuenció un segmento de 532 pb de la región control mitocondrial en 72 individuos, el cual permitió detectar un total de 29 sitios polimórficos, 35 haplotipos, niveles moderados altos de diversidad haplotípica (0.793 – 0.969) y muy bajos niveles de diversidad nucleotídica (0.004 – 0.008). El análisis de flujo génico mostró altos niveles de conectividad entre las poblaciones en las áreas de muestreo. El análisis de varianza molecular (ФST = 0.00868, P = 0.1837), las comparaciones ФST a pares de poblaciones y las pruebas de diferenciación genética confirmaron la carencia de estructuración genética entre las tres localidades. Estos análisis sugieren que los sitios de muestreo analizados pueden ser considerados como un solo grupo genético. El comportamiento migratorio, el alto potencial de dispersión de los estadios tempranos de desarrollo y la ausencia de barreras oceanográficas pueden explicar su homogeneidad genética a lo largo del mar peruano. También se examinó la demografía histórica. Las pruebas de neutralidad, la distribución mismatch y el Bayesian Skyline Plot sugirieron un escenario de expansión poblacional que tuvo lugar durante el Pleistoceno Superior. Este estudio provee información nueva con respecto a la genética de poblaciones de la caballa en el Pacífico Sudeste.
Palabras clave: Pacífico Sudeste; mar peruana; genética de peces; flujo génico; demografía histórica.
Introduction
An essential component of a long-term fisheries management is the identification of population units to implement sustainable fishing strategies (Begg et al. 1999). As fisheries management decisions must be based on accurate knowledge of population structure (Henriques et al. 2012), the determination of the genetic structure of commercial marine fishes is essential (Utter 1994). At present, molecular markers are considered as important tools for this purpose because they offer the possibility of identifying and delineating populations where phenotypic and behavioral characteristics are apparently not enough (Ward 2000). Mitochondrial DNA markers, due to inherent features such as high mutation rate and non-recombinant maternal inheritance, allow investigating several aspects of the structure of fish population (Antoniou & Magoulas 2013). It has been shown that the control region, considered the most variable fragment of the mitochondrial genome (Meyer 1993), is useful for detecting adequate levels of genetic differentiation in marine fish (Viñas et al. 2004, Wang et al. 2008).
The present-day genetic structure of marine fishes commonly reflects the combination of historical events, contemporary conditions and life-history traits (Fauvelot & Planes 2002). The complex interactions between dispersal ability of larva and adults and present-day barriers such as oceanographic currents, upwelling cells, sea temperature and isolation by distance have been responsible for the patterns of genetic variation in marine populations (Hemmer-Hansen et al. 2007). Pleistocene ice ages and consequent eustatic sea level oscillations have also greatly influenced the distribution and population sizes of temperate marine species. Under these fluctuating paleoclimatic conditions, consequences on the both current genetic diversity and population structure can be expected (Hewitt 2004).
The Peruvian marine ecosystem comprises the Northern segment of the Humboldt Current System (hereinafter NHCS). Equatorward cold-water Peru Coastal Current (PCC), strong temporal variability (e.g. El Niño Southern Oscillation), permanent and intense wind-driven coastal upwelling and the greater global fishing productivity are the most outstanding features of this ecosystem (Bakun & Weeks 2008, Chavez et al. 2008). Here the chub mackerel Scomber japonicus Houttuyn 1782 (Perciformes: Scombridae) is a pelagic fish of high economic importance that records the best historical landings after the Peruvian anchovy Engraulis ringens and the Chilean jack mackerel Trachurus murphyi (Montecino & Lange 2009). Due to its high biomass and meat quality (e.g. fatty acids) (NOAA 2015), it supports a significant export-based economic activity. The chub mackerel is currently the fifth most exploited species of pelagic fish worldwide (FAO 2016). This middle-sized scombrid is restricted on the continental shelves of tropical and subtropical regions of the Pacific Ocean. Within the Southeast Pacific it is distributed from Panama to Chile (45°41’S) including Cocos, Malpelo and Galapagos Islands (Collette et al. 2011). Off Peru, the chub mackerel is mainly associated with the water mass denominated Subtropical Surface Waters (SSW) (Sanchez et al. 2000) usually forming large shoals especially beyond 160 nautical miles (Dioses 1995). This species is considered as an opportunistic omnivorous being the Peruvian anchovy, copepods and euphausiids their main prey (Alegre et al. 2015). Because larger fishes, marine mammals and seabirds consume it, S. japonicus links lower trophic levels with top-predators, making this species play an important role in the NHCS trophic web. Its distribution and abundance depends on both prey availability and the environmental variability of the NHCS (Gutiérrez et al. 2012). During warm episodes such as summers and El Niño Events, SSW coastward intrusions cause the species to approach the coast, making it more vulnerable to purse seiners (Gutiérrez et al. 2012). In fact, the greatest historic landing in Peru was during the very strong 1997-98 El Niño event (Ñiquen & Bouchon 2004). However, its biomass collapsed after this warm episode and catches have fluctuated since then. At present, chub mackerel landings in Peru are reported as an important fishing associated mainly with catches of Chilean jack mackerel (Gutiérrez et al. 2012).
In the Northwest Pacific, where the chub mackerel is of great importance, both mitochondrial and microsatellite markers have been used to reveal its genetic structure. Based on mitochondrial control region, no genetic differentiation was found among three sampling sites off Taiwan (Tzeng et al. 2007). A moderate level of genetic differentiation was observed among East and South China Seas based on eleven microsatellite loci, proposing two divergent stocks (Zeng et al. 2012). A more extensive study ranging from the Yellow to South China Sea based on fifteen microsatellite loci found three moderately differentiated genetic groups (Cheng et al. 2014). Based on mitochondrial cytochrome b sequences, a moderate genetic differentiation was also found between South and East China Seas, which was attributed to sea level fluctuations during Late Pleistocene (Zhu et al. 2014). However, based on eight microsatellite loci, a genetic homogeneity was observed between samples from the South and East China Seas, coupled to weak but significant genetic structure between samples of China and Japan (Cheng et al. 2015). Using sequences of the mitochondrial control region, no genetic differentiation was found along the Chinese coast, but a significant structure was observed between populations of China and Japan, which was attributed to an isolation during Late Pleistocene (Yan et al. 2015).
In the Southeast Pacific, two previous non-molecular studies based on spawning areas (Serra 1982) and growth parameters (Dawson 1986) have suggested the presence of two chub mackerel stocks: "Ecuador - Northern Peru" and "Southern Peru - Northern Chile". Despite its high economic and ecological importance, it is practically unknown its population genetics in the Southeast Pacific and there are no molecular studies that contrast these hypotheses. Nowadays, the generation of information based on molecular markers is considered substantial for sustainable use and effective management of fisheries (Ward 2000). Understanding the population genetics of S. japonicus will be useful for the development of sustainable fishing strategies in order to avoid future potential collapses. In this study, we sequenced a 532 bp segment of the mitochondrial control region from 72 Peruvian samples from the Peruvian coast. The first goal was to estimate the levels of genetic structure and gene flow. The hypothesis of a single panmictic unit due to its high dispersal potential and the apparent lack of oceanographic barriers in the NHCS was tested in opposition to the two previous two-stock hypotheses. The second goal was to check its population history using statistical tests. We hypothesized that Pleistocene climatic oscillations and their characteristics life-history traits influenced significantly on its historical demography. The results indicate that chub mackerel lacks genetic structure, exhibits high levels of gene flow and that it passed through a population expansion that took place during Late Pleistocene.
Material and methods
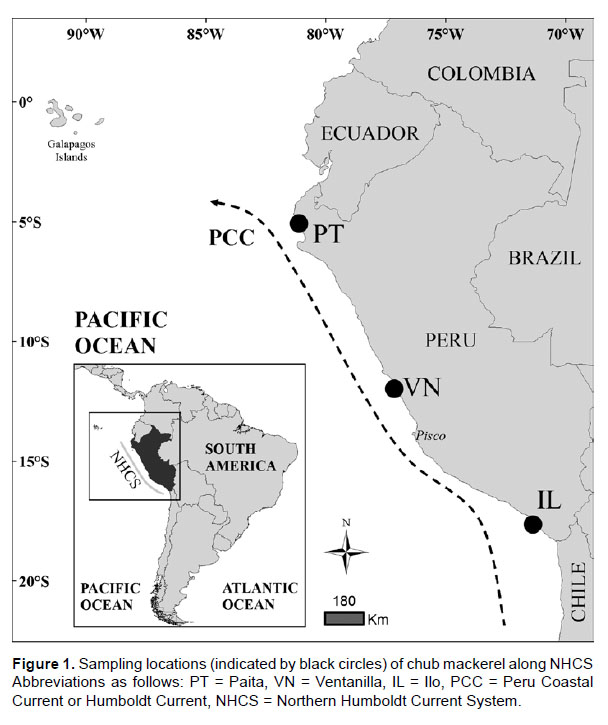
Sampling and DNA sequencing.- Chub mackerel individuals were captured by purse seiners during the summers of years 2013-2014 in coastal waters of the Peruvian coast (Fig. 1, Table 1). Taxonomic identification was carried out using morphological keys. A small skeletal dorsal muscle fragment was excised from each individual and preserved in 100% ethanol at 4 °C. Genomic DNA was obtained using a modified salt-extraction protocol (Aljanabi & Martinez 1997), checked with 1% agarose gel electrophoresis and quantified by NanoDrop® Spectrophotometer (Thermo Fisher Scientific Inc.). A 532 bp segment of the mitochondrial control region of S. japonicus was amplified by Polymerase Chain Reaction (PCR) with the primers L-Serg 5’-CTCAGCGTTTAGAGCATCGGTC-3’ (forward primer, designed in this study based on the complete mitogenome of S. japonicus, accession number: AB488405) and HARN12S 5’-GGGCTGGCACGAGTTTTACCG-3’ (reverse primer, Ore-Chavez pers. comm.). PCR reactions were performed in a volume of 50 µL containing 2.5 mM MgCl2, 1X PCR Buffer, 0.2 µM of each primer, 0.2 mM of each dNTP, 0.5 U Taq Polymerase (InvitrogenTM, USA) and 20 ng of template DNA. We carried out amplifications in an Applied Biosystems GeneAmp® PCR System 9700 Thermal Cycler using the following thermal profile: 5 min of initial denaturing step (94 °C), 35 cycles of denaturing (94 °C, 1 min), annealing (55 °C, 1 min) and extension (72 °C, 2 min), and a final extension step of 72 °C (10 min). The PCR products were shipped to be purified and sequenced by Macrogen Inc. (Maryland, USA).
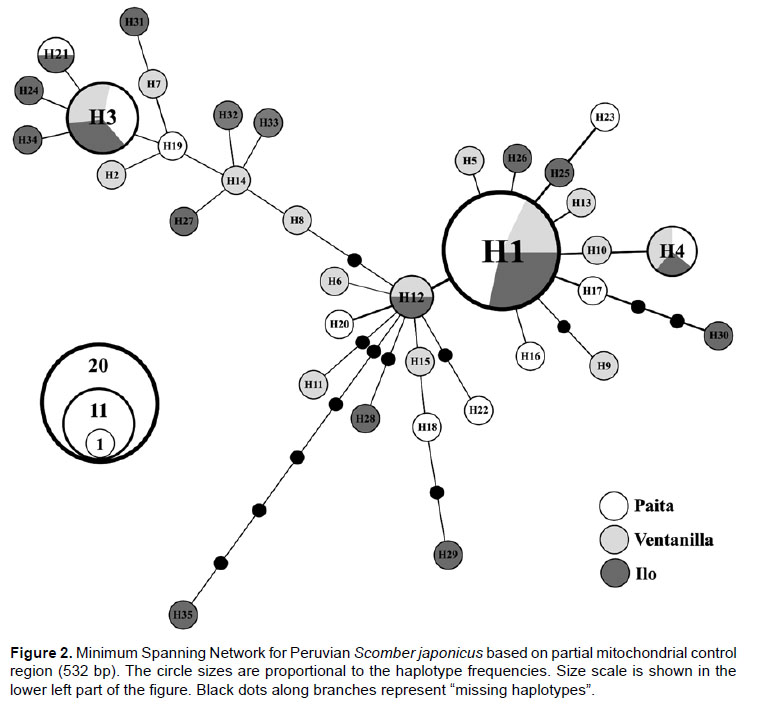
Data Analysis.- DNA sequences were verified by visual examination and aligned using Bioedit Sequence Alignment Editor 7.0.5.3 (Hall 1999). Number of haplotypes (H), number of polymorphic sites (S), haplotype diversity (Hd) and nucleotide diversity (π) were calculated using Arlequin 3.5 (Excoffier & Lischer 2010). A Minimum Spanning Network (MSN) was constructed with HapStar (Teacher & Griffiths 2011) using distances computed in Arlequin in order to visualize the genealogical relationships among haplotypes.
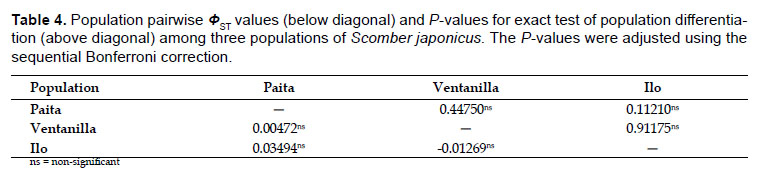
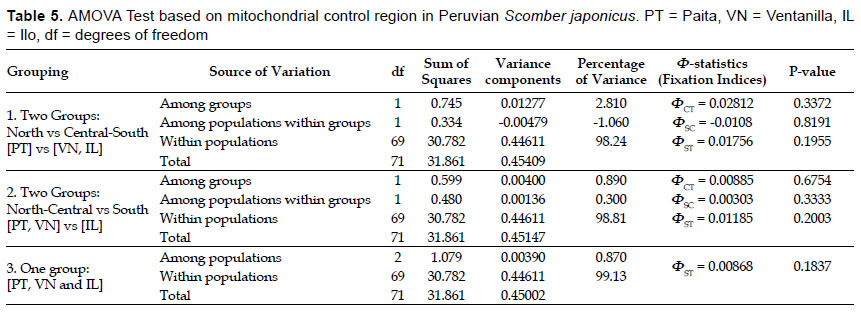
Analysis of molecular variance (Excoffier et al. 1992) was applied to evaluate the distribution of the genetic variation among sampling locations. The products of an AMOVA consist in variance components estimations and Ф-statistics (Analogues to Wright’s FST) associated to three population hierarchies: ФCT (among groups), ФSC (among populations within regions) and ФST (within populations). Three user-defined structures were evaluated and significance levels were computed using 10,000 non-parametric permutations. Pairwise ФST comparisons also were performed to estimate genetic differentiation among populations. The null hypothesis of panmixia (random distribution of haplotypes among populations) was also tested using an exact test of population differentiation (Raymond & Rousset 1995). Probabilities (p-values) were estimated using 10000 non-parametric permutations. These tests were carried out in Arlequin. Probabilities were estimated using 10000 nonparametric permutations. The P-values were adjusted using the sequential Bonferroni correction (Rice 1989).
Statistics of genetic differentiation HST and KST(Hudson et al. 1992) were performed for testing the null hypothesis of sampling populations are not genetically different to each other. Significance levels were calculated with 1000 permutation replicates. Three Wright’s FST analogues measures of population differentiation for DNA sequences were calculated: the haplotype-based index GST (Nei 1973) and the sequence-based indexes NST (Lynch & Crease 1990) and FST (Hudson et al. 1992. Gene flow estimates (Nm), i.e. expected number of migrant females between populations per generation, were derived from these indexes based on island model. These statistical tests and indexes were carried out in DnaSP v5 (Librado & Rozas 2009). Furthermore, migration rates (M) in each direction between each pair of sample location were estimated under a Bayesian MCMC coalescent-based approach with MIGRATE-n v. 3.6.4. (Beerli 2006).
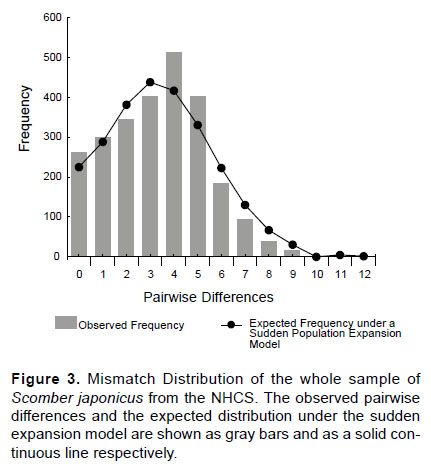
Historical demography of the chub mackerel was inferred using several approaches. First, the mismatch distribution analysis (Roger & Harpending 1992) was applied using Arlequin 3.5. Mismatch distribution compares observed frequencies of pairwise differences with a theoretical distribution under the sudden population expansion model. A multimodal distribution indicates a long-term population stability while a smooth distribution is commonly related to past collapse followed by a sudden expansion (Rogers & Harpending 1992). Harpending’s raggedness index (Hri) and Sum of Squared deviations (SSD) were included to determine the fit to the expected curve under the sudden population expansion model. Hri quantifies the smoothness of the observed mismatch distribution. Low and non-significant Hri values usually indicate an expanded population while a significant Hri (P < 0.05) is evidence for rejecting expansion model. Computation and significance estimation of both goodness of fit tests was assessed using 10000 permutations in Arlequin. Moment estimator of time of expansion (tau) and the past demographic parameters before (θ0) and after expansion (θ1) were determined and expressed in units of mutational time (Rogers & Harpending 1992) using Arlequin. To estimate the date of population expansion the equation t = tau/2u was used, where t = date of the growth in generations, u is the cumulative mutation rate per generation of the whole DNA region under study which is calculated as u = 2µk (µ is the mutation rate per nucleotide and k is the nucleotide length of analyzed marker) (Rogers & Harpending 1992). Owing to that the molecular clock for the mitochondrial control region of fishes seems to vary among different taxa and the lack of scombrid fossil data, a previous used mutation rate for S. japonicus (11 – 16%MY-1) was adopted (Yan et al 2015). To calculate the expansion time, a mean generation time of 2 years for S. japonicus was used (Tzeng et al. 2007).
Fu’s Fs (Fu 1997) and R2 statistics were used to examine deviations from mutation-derive equilibrium (neutral theory model). Both tests are considered as the most powerful for detecting population growth. Fu’s Fs is best for large sample sizes while R2 is the most suited statistics when sample size is small (Ramos-Onsins & Rozas 2002). Significant-negative Fu’s Fs and significant-positive R2 values suggest rejection of neutrality and can be interpreted as signatures of population expansion. Significances of both neutrality tests were evaluated with 10 000 coalescent simulations in Arlequin and DnaSP v5 respectively. Historical trends in effective population size were investigated using Bayesian Skyline Plot (BSP) as implemented in BEAST v 1.6.1 (Drummond & Rambaut 2007). A strict molecular clock model (mutation rate = 16% MY-1) and the best-fit substitution model suggested by jModeltest (Posada, 2008) were selected. The number of grouped intervals was set to 10 and the BSP was performed in the piecewise-constant model. The BSP was plotted in Tracer v1.5 (Rambaut & Drummond 2009).
Results
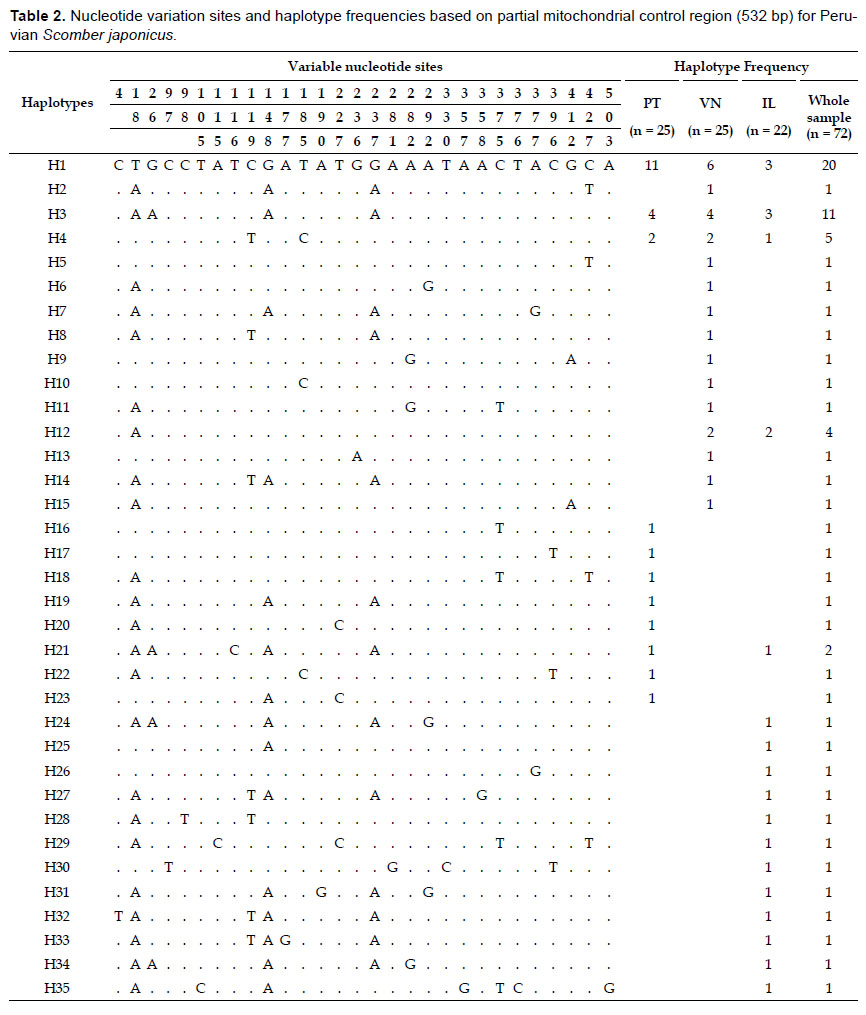
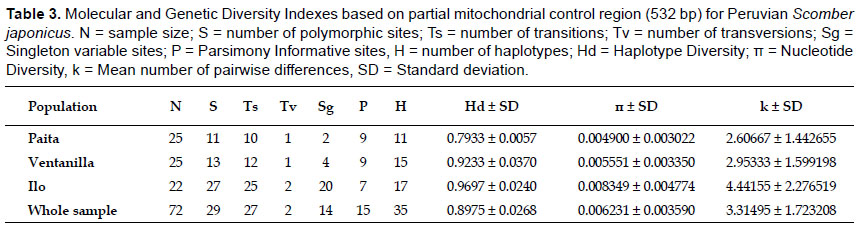
Genetic diversity and network.- A 532 bp fragment of the mitochondrial control region was obtained from 72 individuals belonging three fishing areas of the Peruvian coast. Sequences were deposited in GenBank database under accession numbers KT696367 - KT696438. We detected 29 polymorphic sites including 27 transitions and two transversions, giving rise 35 haplotypes (Table 2). No indels were found. The most common haplotype (H1) was shared by three populations alongside two other less common ones (H3 and H4). Two populations shared haplotypes H12 and H21 while all other remaining haplotypes were population-specific (Table 2). In general, haplotype diversities detected were moderate to high (overall = 0.8975) while nucleotide diversity values were very low (overall = 0.006231) (Table 3). The southern sampling location (Ilo) showed the highest values of both haplotype (0.9697) and nucleotide (0.008349) diversities. The Minimum Spanning Network displayed an atypical star-like topology. No clear central haplotype was detected, but there are two main haplogroups with H1 and H3 as ancestral haplotypes that can be visualized. The most haplotypes differ in a single mutation step, except for some divergent ones (e.g. H30 and H35) (Fig. 2). In general, concordance between haplotypes and geographic sampling sites cannot be appreciated.
Population structure and Gene Flow.- No significant genetic structure was observed on the Peruvian coast based on population pairwise comparisons (Table 4). The same conclusion was also supported by the AMOVA test (Table 5). The three groupings tested in AMOVA indicated that most of the genetic variation was within populations (~98%) with very low and non-significant ΦST indexes (ΦST = 0.008 – 0.03, P > 0.05). Variation among populations was also non-significant. Very low variances and non-significant fixation index at amonggroups’ hierarchy were detected (ΦCT = 0.008 – 0.02, P < 0.05). The exact test of population differentiation showed non-significant P-values among sampling locations (Table 4) so the null hypothesis of panmixia was not rejected. Genetic differentiation estimates for whole sample based on haplotype-based statistic (HST = 0.00603, P = 0.193) and sequence-based statistic (KST = 0.00822, P = 0.18) were very low and non-significant, so the three locations are practically non-genetically different. Wright’s FST analogues population differentiation measures and their respective Nm values were congruent with very low levels of genetic differentiation and very high levels of gene flow (GST = 0.00569, Nm = 87.34; NST = 0.01291, Nm = 38.23; FST = 0.01298, Nm = 38.02). Gene flow was also estimated through pairwise migration rates using a coalescent method as implemented in MIGRATE software. Migration rates show bidirectional and asymmetric movements between sampling populations. Overall, southward migration rates were higher than northward migration rates (MPT→VN=866.1, MPT→IL= 865.1, MVN→IL= 589.1). Only one northward migration rate was strong (MIL→VN= 638) while the others were basically weak (MVN→PT= 195.4, M IL→PT= 410.1).
Historical demography.- The mismatch distribution curve for the whole sample showed a clearly unimodal pattern (Fig. 3). Hri and SSD showed low and non-significant values (Hri = 0.01393, P > 0.05; SSD = 0.00344, P > 0.05) indicating a good fit, so the expansion model was not rejected. The results of Fu’s Fs test showed large negative and very significant values, indicating an excess of rare haplotypes over what would be expected under neutrality. R2 statistic showed positive values very close to the significance (Table 6). The position of the mismatch distribution crest, which reflects the time of the population growth, is determined by tau parameter. For the whole sample, tau resulted 4.168 (Table 6), therefore the population growth is estimated that took place 4.168/2u generations ago.
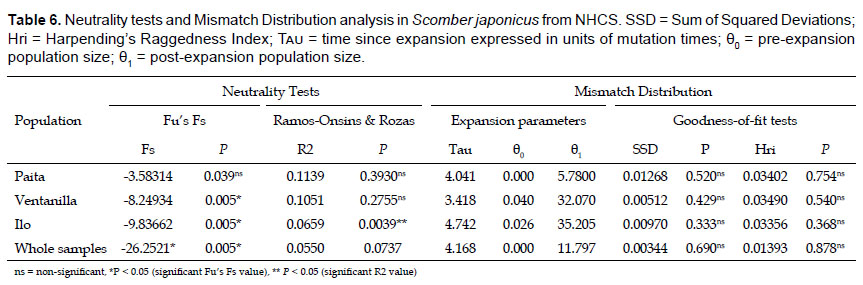
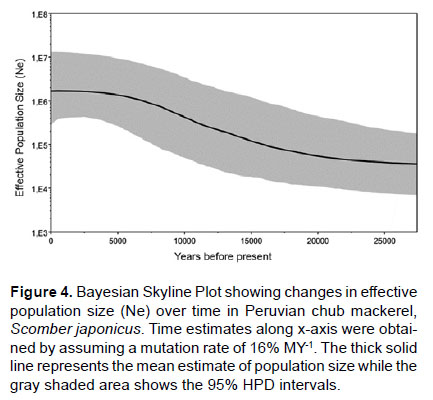
Give a mutation rate of 11 – 16% MY-1 (Yan et al. 2015) the population expansion of the chub mackerel in the NHCS was estimated to have taken place 35.611 to 24,483 years before present. Hasegawa-Kishino-Yano (HKY) model was selected and a mutation rate of 16% MY-1 were used for the Bayesian Skyline plot. BSP revealed a population growth that started around 25 thousand years before present, which reflected approximately the estimates of time since expansion calculated with Rogers & Harpending’s formula (Fig. 4).
Discussion
Studies of molecular genetics in marine resources of the Humboldt Current System (HCS), one the most productive fishing regions in the world, are scarce. In the case of the chub mackerel, research that provides genetic information for fisheries management purposes in this region has not been reported to date. Data from mitochondrial sequences will be useful for developing long-term sustainable fishing strategies. In addition, historical demography analysis will allow evaluating Pleistocene climate-driven impacts in this sensitive species. These issues were addressed by combining the information obtained from mitochondrial control region sequences along with specific statistical tests.
According to the results, the chub mackerel does not show genetic structuring in its Peruvian distribution. Analysis of molecular variance, exact population differentiation tests and ΦST pairwise comparisons suggest a lack of population differentiation. Besides, since there is no obvious haplotype-geography association in the minimum spanning network a genetic structure cannot be appreciated. This finding is consistent with the assumption of marine pelagic fish species with dispersal capabilities and large effective population sizes generally show low genetic differentiation among populations (Palumbi 1994, Ward et al. 1994). Some chub mackerel’s characteristic lifehistory traits such as high larval dispersal capacity, schooling behavior, high fecundity and seasonal spawning migrations (Castro & Santana 2000) may explain its genetic homogeneity in the NHCS. Similar results has also been reported in other chub mackerel populations. A study carried out in Taiwan reported a clearly lack of genetic structure among three sampling locations (Tzeng et al. 2007), while a recent study found a lack of genetic structure along the coast of China suggesting a high level of gene flow (Yan et al. 2015). The blue mackerel Scomber australasicus of Taiwan also showed a lack of genetic heterogeneity (Tzeng 2007). This genetic homogeneity seems also obey the apparent lack of major oceanographic barriers within the NHCS. The area off Pisco (~14°S) is the main Peruvian upwelling zone (Gutiérrez et al. 2011). Pisco appear to be the delimitation between the two previously proposed chub mackerel stocks (Dawson 1986, Serra 1982). Strong upwelling cells are believed to be responsible for gene flow disruption between adjacent marine populations (Waters & Roy 2004). For example, a genetic break found in the sciaenid Atractoscion aequidens throughout the Benguela Current Region highlights the important role of perennial Lüderitz upwelling cell in the connectivity of this species (Henriques et al. 2014). However, no genetic differentiation signals along the Peruvian coast in S. japonicus was observed, which means that strong Pisco upwelling cell is not an effective barrier to gene flow of S. japonicus. In addition, the lack of other major surface coastal currents and oceanographic barriers in the NHCS probably makes this marine environment has a genetic homogenizing effect.
Estimation of migration rates suggests a bidirectional and asymmetric historic gene flow with an important movement toward the south. Reproductive migrations may explain this behavior. It is known that S. japonicus spawns from 03°30’S to 18°30’S with preference in the summers when the SSWs approach the coast, but it is believed that the waters of the northcentral region of Peru are the main spawning point(Santander & Flores 1983). This fact may explain the high gene flow between central (VN) and northern (PT) sampling locations. However, a strong northward gene flow since the southern location (IL) to Ventanilla was detected. Several rare haplotypes resulting from Ilo’s exclusive mutations, i.e. singletons, cause that the genetic diversity of S. japonicus at here is greater compared to the other two localities(Table 3). This observed decreasing-northward haplotype diversity along the Peruvian coast can be considered a possible signal of an important northward migration as a consequence of a potential and non-reported spawning grounds off Ilo, or an important migration from Chilean waters. Another major factor is the passive displacement of eggs and larvae dispersed toward the north by the Peruvian Coastal Current (PCC) prior to metamorphosis. However, feeding movements also can explain bidirectional migration. It is largely known that the distribution and abundance of S. japonicus depend on a large extent on food availability. It can migrate through both neritic and epipelagic environments usually forming interspecific schools with the Peruvian anchovy to feed. Summarizing, reproductive and feeding migrations and passive transport of eggs and larvae would allow mixing of genes in an asymmetric bidirectional way along the Peruvian sea.
The overall haplotype diversity found in Peruvian chub mackerel (Hd = 0.8975) is lower than mean values found in other control region-based studies such as Taiwan (Hd = 0.971) (Tzeng et al. 2007), China (Hd = 0.958) (Yan et al. 2015) and Atlantic Ocean (Hd = 0.98, population currently recognized as the species Scomber colias) (Zardoya et al. 2004). On the other hand, the most of studies employing mitochondrial control region sequences to investigate genetic structuring of exploited marine fishes have observed values of π much higher than those recorded for S. japonicus in the present work. Nucleotide diversities (π = 0.0049 – 0.00835) found in this study were ten times lower than other chub mackerel populations: 0.0171 (Tzeng et al. 2007), 0.01074 (Yan et al. 2015), and very similar to those reported for Taiwanese Scomber australasicus (π = 0.007) (Tzeng 2007), the overexploited Brazilian sciaenid Cynoscian acoupa (π = 0.00288) (Rodrigues et al. 2008) and the highly exploited Chilean jack mackerel Trachurus murphyi (π = 0.0006) (Cárdenas et al. 2009). The moderated Hd values and very low π values in Peruvian chub mackerel may be reflecting the oceanographic features of NHCS, such as strong permanent upwelling and ENSO events which periodically "resets" the ecosystem (Bakun & Weeks 2008). However, harmful anthropogenic activities like overfishing may also generate a pressure on the genetic diversity in marine fishes (Rodrigues et al. 2008). Interestingly, the highest genetic diversities detected in this study were found in Ilo, where this species is subject to less fishing pressure (Gutiérrez et al. 2012). A continuous genetic sampling at different temporal scales would help give clues to test this hypothesis.
Based on the combination of haplotype and nucleotide diversity, four basic scenarios for marine fish have been proposed for marine fishes (Grant & Bowen 1998). In this study, a combination of moderate values of haplotype diversity coupled with very low values of nucleotide diversity was detected in Peruvian chub mackerel. This situation can be considered consistent with the scenario of population expansion after a period of low effective population size. Unimodal mismatch distribution coupled with the rejection of neutrality clearly suggest a scenario of ancestral population expansion, which is supported by the Bayesian Skyline Plot. The occurrence of star-like patterns radiating from ancestral haplotypes suggesting a population expansion after a bottleneck also support this finding. Timing of expansion by tau parameter estimation was about 35611 to 24483 years before present, time closely coincident with the Last Glacial Maximum (LGM). This important climatic event during the Late Pleistocene may have had a great effect on the historical demography of the chub mackerel (Hewitt 2004). LGM was characterized by large ice sheet extensions, strong marine regression and changes in oceanographic patterns and shorelines. An abrupt decline in sea level (about 120 – 140m below current sea level) induced a massive exposure of the continental shelf (Lambeck et al. 2002). The chub mackerel is considered a sensitive species to environmental changes (NOAA 2015). Habitat loss due to the exposition of continental shelf caused by the LGM sea-level lowstand coupled with the hard paleo-oceanographical conditions (changes in salinity, temperature and nutrition levels) likely caused a population decline, at least the Peruvian part of its distribution, and surviving individuals could be adapted in glacial refugia. This is consistent with the minimum abundances of fishes in the LGM inferred by paleoproductivity proxies (scales and bones) in laminated sediments of the Peruvian continental shelf (Salvatteci 2013). Then, at the beginning of Holocene, post-glacial sea-level rise and increasing of primary productivity off Peru (Rein et al. 2005) may have contributed to the recuperation of habitat and the consequent recolonization on surrounding areas. Similar historical demography patterns were found in the chub mackerel from Taiwan (Tzeng et al. 2007), China (Yan et al. 2015, Zhu et al. 2014) and other scombrids of other latitudes (Ely et al. 2005, Theisen et al. 2008, Tzeng 2007). Molecular genetic studies in pelagic fishery resources of the Humboldt Current System such as jack mackerel Trachurus murphyi (Cárdenas et al. 2009), squid Doryteuthis gahi (Ibañez et al. 2012) and jumbo squid Dosidicus gigas (Ibañez et al. 2011, Sanchez et al. 2016) have shown similar population histories. Despite of these species have different life-history traits, it is feasible to recognize that Pleistocene climatic oscillations are among the most influential factors in the historical demography of marine resources in the Humboldt Current System.
Since a great deal of genetic data for the detection of fish populations is required, is necessary to acknowledge the inherent limitations of this study. The sampling effort probably does not represent the large effective population size of this species. One consequence of a small sample size is that the detection of haplotypes may have been underestimated resulting in missing haplotypes and a weak signal of matrilineal lineages. Besides, mitochondrial DNA has some known disadvantages (Ballard & Whitlock 2004). Despite these limitations, the results in this study appear to be robust regarding genetic homogeneity detected. However, further studies will require more extensive sampling by increasing collection sites and sample sizes. It is also necessary to conduct studies using nuclear markers with a multilocus approach. For example, the use of several microsatellite loci available for this species (Yagishita & Kobayashi 2008, Cha et al 2010, Zeng & Cheng 2012) would allow a better resolution for the detection of population units in the Southeast Pacific, as it has been done in the Northwest Pacific (Zeng et al 2012, Cheng et al 2014, Cheng et al 2015). The creation of time series based on a panel of microsatellites at the same sampling sites to monitor changes in allele frequencies and to estimate effective population sizes (Ne) would be very informative for management purposes, e.g. to manage stocks and avoid overfishing. Given the potential existence of shared stocks between Chile, Peru and Ecuador a comprehensive analysis based on molecular markers in its full geographic range is highly required for fisheries management purposes.
This study provides the first snapshot view regarding the population genetics of S. japonicus in the Southeast Pacific. This paper is an initial effort for generating knowledge for fisheries management purposes in this economically important species. In conclusion, based on our genetic analysis, the single panmictic unit hypothesis is supported. Mitochondrial control region sequences left clear signals that support the hypothesis of important demographic events during Late Pleistocene. Finally, due to the moderate haplotype diversity and mainly by the very low nucleotide diversity found here, monitoring and regulation measures are urgently needed for the fishery of this species in Peruvian sea.
Acknowledgments
We thank Walter Condori and Javier Villanueva (IMARPE Headquarter Ilo) for their assistance in the sample collection and preparation. We also would like to thank to LEMBA colleagues Diego Deville and Paola Castro for their help in the lab and Ximena Velez-Zuazo for the invaluable and constructive insights. Special acknowledgments to the editor and anonymous reviewers for their comments, which greatly improved the final version of this manuscript. This work has been supported by a funding from the Research Vicerrectorate of Universidad Nacional Mayor de San Marcos (VRI-UNMSM Code 141001027).
Literature cited
Alegre A., A. Bertrand, M. Espino, et al. 2015. Diet diversity of jack and chub mackerels and ecosystem changes in the northern Humboldt Current system: A long-term study. Progressin Oceanography 299–313. http://doi.org/10.1016/j.pocean.2015.07.010 [ Links ]
Aljanabi S.M. & I. Martinez. 1997. Universal and rapid salt-extractionof high quality genomic DNA for PCR-based techniques.Nucleic Acids Research 25(22), 4692–4693. http://doi.org/10.1093/nar/25.22.4692 [ Links ]
Antoniou A. & A. Magoulas. 2013. Application of Mitochondrial DNA in Stock Identification. In S. X. Cadrin, L. A. Kerr, & S. Mariani (Eds.), Stock Identification Methods: Applications in Fishery Science (2nd ed., pp. 257–295).Academic Press. http://doi.org/10.1016/B978-0-12397003-9.00013-8 [ Links ]
Bakun A. & S.J. Weeks. 2008. The marine ecosystem off Peru: What are the secrets of its fishery productivity and what might its future hold? Progress in Oceanography 79(2–4), 290–299. http://doi.org/10.1016/j.pocean.2008.10.027 [ Links ]
Ballard J.W.O. & M.C. Whitlock. 2004. The incomplete naturalhistory of mitochondria. Molecular Ecology 13, 729–744. http://doi.org/10.1046/j.1365-294X.2003.02063.x [ Links ]
Beerli P. 2006. Comparison of Bayesian and maximum-likelihoodinference of population genetic parameters. Bioinformatics,22(3), 341–345. http://doi.org/10.1093/bioinformatics/bti803 [ Links ]
Begg G.A., K.D. Friedland & J.B. Pearce. 1999. Stock identification and its role in stock assessment and fisheries management: An overview. Fisheries Research 43, 1–8. http://doi.org/10.1016/S0165-7836(99)00062-4 [ Links ]
Cárdenas L., A.X. Silva, A. Magoulas, et al. 2009. Genetic population structure in the Chilean jack mackerel, Trachurus murphyi (Nichols) across the South-eastern Pacific Ocean. Fisheries Research 100(2), 109–115. http://doi.org/10.1016/j.fishres.2009.06.013 [ Links ]
Castro J.J. & A.T. Santana. 2000. Synopsis of biological data on the Chub Mackerel (Scomber japonicus Houttuyn, 1782). FAOFisheries Synopsis, 75(157). [ Links ]
Cha, H. K., H.S. An, J.H. Choi, S. Kang, J. Park, & K. Kim. 2010. Isolation and characterization of polymorphic microsatellitemarkers for genetic analysis of chub mackerel (Scomberjaponicus). Conservation Genetics Resources 2(1), 7–9.https://doi.org/10.1007/s12686-009-9123-7 [ Links ]
Chavez F.P., A. Bertrand, R. Guevara-Carrasco, et al. 2008. The northern Humboldt Current System: Brief history, present status and a view towards the future. Progress in Oceanography 79(2–4), 95–105. http://doi.org/10.1016/j. pocean.2008.10.012 [ Links ]
Cheng J., T. Yanagimoto, N. Song & T. Gao. 2015. Populationgenetic structure of chub mackerel Scomber japonicusin the Northwestern Pacific inferred from microsatellite analysis. Molecular Biology Reports 42, 373–382. http://doi.org/10.1007/s11033-014-3777-2 [ Links ]
Cheng Q., Y. Zhu & X. Chen. 2014. High polymorphism andmoderate differentiation of chub mackerel, Scomber japonicus (Perciformes: Scombridae), along the coast of China revealed by fifteen novel microsatellite markers. Conservation Genetics 15(5), 1021–1035. http://doi.org/10.1007/s10592-014-0596-x [ Links ]
Collette B.B., A. Acero, C. Canales, et al. 2011. Scomber japonicus. The IUCN Red List of Threatened Species 2011:e.T170306A6737373. [ Links ]
Dawson W.A. 1986. The interpretation of otolith structure for theassessment of age and growth of some pelagic fishes fromthe coast of Ecuador, Mackerel (Scomber japonicus),pacific sardine (Sardinops sagax), pacific thread herrings(Opisthonema medirastre and Opisthonema bulleri),and round herring (Etrumeus teres). Boletin Cientifico yTecnico, Instituto Nacional de Pesca, Guayaquil - Ecuador9(1), 1–24. [ Links ]
Dioses T. 1995. Análisis de la distribución y abundancia de los recursosjurel y caballa frente a la costa peruana. IMARPE, 3, 1–55. [ Links ]
Drummond A.J. & A. Rambaut. 2007. BEAST: Bayesian evolutionaryanalysis by sampling trees. BMC Evolutionary Biology 7(1), 214. http://doi.org/10.1186/1471-2148-7-214 [ Links ]
Ely B., J. Viñas, J.R. Alvarado Bremer, D. Black, L. Lucas, K. Covello, A.V. Labrie & E. Thelen. 2005. Consequences of thehistorical demography on the global population structureof two highly migratory cosmopolitan marine fishes: theyellowfin tuna (Thunnus albacares) and the skipjack tuna(Katsuwonus pelamis). BMC Evolutionary Biology 5, 19.http://doi.org/10.1186/1471-2148-5-19 [ Links ]
Excoffier L. & H.E.L. Lischer. 2010. Arlequin suite ver 3.5: anew series of programs to perform population geneticsanalyses under Linux and Windows. Molecular EcologyResources 10(3), 564–567. http://doi.org/10.1111/j.17550998.2010.02847.x [ Links ]
Excoffier L., P.E. Smouse, J.M. Quattro. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restrictiondata. Genetics, 131(2), 479–491. [ Links ]
FAO (Food and Agriculture Organization). 2016. The State of World Fisheries and Aquaculture 2016. Contributing to foodsecurity and nutrition for all. Rome. [ Links ]
Fauvelot C., & S. Planes. 2002. Understanding origins of present-day genetic structure in marine fish: Biologically or historically driven patterns? Marine Biology 141(4), 773–788. http://doi.org/10.1007/s00227-002-0869-7 [ Links ]
Fu Y.X. 1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection.Genetics, 147(2), 915–925. [ Links ]
Grant W.S. & B.W. Bowen. 1998. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. Journal of Heredity 89(5), 415–426. http://doi.org/10.1093/jhered/89.5.415 [ Links ]
Gutiérrez D., I. Bouloubassi, A. Sifeddine, et al. 2011. Coastal coolingand increased productivity in the main upwelling zone offPeru since the mid-twentieth century. Geophysical ResearchLetters, 38(7), 1–6. http://doi.org/10.1029/2010GL046324 [ Links ]
Gutiérrez M., R. Castillo, M. Segura, et al. 2012. Trends in spatiotemporal distribution of Peruvian anchovy and other small pelagic fish biomass from 1966-2009. Latin AmericanJournal of Aquatic Research 40(3), 633–648. http://doi.org/103856/vol40-issue3-fulltext-12 [ Links ]
Hall T.A. 1999. BioEdit: a user-friendly biological sequence alignmenteditor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95-98. [ Links ]
Hemmer-Hansen J., E.E. Nielsen, P. Grønkjær & V. Loeschcke. 2007.Evolutionary mechanisms shaping the genetic population structure of marine fishes; lessons from the European flounder (Platichthys flesus L.). Molecular Ecology 16(15), 3104–3118. http://doi.org/10.1111/j.1365-294X.2007.03367.x [ Links ]
Henriques R., W.M. Potts, C.V Santos, et al. 2014. Population connectivity and phylogeography of a coastal fish, Atractoscion aequidens (Sciaenidae), across the Benguela Current region:Evidence of an ancient vicariant event. PLoS ONE, 9(2). http://doi.org/10.1371/journal.pone.0087907 [ Links ]
Henriques R., W.M. Potts, W.H.H. Sauer & P.W Shaw. 2012. Evidence of deep genetic divergence between populations of an important recreational fishery species, Lichia amia L.1758, around southern Africa. African Journal of Marine Science 34(4), 585–591. http://doi.org/10.2989/1814232X.2012.749809 [ Links ]
Hewitt G.M. 2004. Genetic consequences of climatic oscillations in the Quaternary. Philosophical Transactions of the RoyalSociety B: Biological Sciences 359(1442), 183–195. http://doi.org/10.1098/rstb.2003.1388 [ Links ]
Hudson R.R., M. Slatkin & W.P. Maddison. 1992. Estimation of levels of gene flow from DNA sequence data. Genetics,132(2), 583–589. [ Links ]
Ibañez C.M., L.A. Cubillos, R. Tafur, et al. 2011. Genetic diversity and demographic history of Dosidicus gigas (Cephalopoda: Ommastrephidae) in the Humboldt Current System. MarineEcology Progress Series (MEPS), 431, 163–171. http://doi.org/10.3354/meps09133 [ Links ]
Ibañez C.M., J. Argüelles, C. Yamashiro, et al. 2012. Spatial genetic structure and demographic inference of the Patagoniansquid Doryteuthis gahi in the south-eastern Pacific Ocean. Journal of the Marine Biological Association of the United Kingdom, 92(1), 197–203. http://doi.org/10.1017/S0025315411000440 [ Links ]
Lambeck K., T.M. Esat & E.K. Potter. 2002. Links between climate and sea levels for the past three million years. Nature, 419(6903),199–206. http://doi.org/10.1038/nature01089 [ Links ]
Librado P. & J. Rozas. 2009. DnaSP v5: A software for comprehensiveanalysis of DNA polymorphism data. Bioinformatics, 25(11),1451–1452. http://doi.org/10.1093/bioinformatics/btp187 [ Links ]
Lynch M. & T.J. Crease. 1990. The analysis of population surveydata on DNA sequence variation. Molecular Biology and Evolution, 7(4), 377–394 [ Links ]
Meyer A. 1993. Evolution of mitochondrial DNA in fishes. In P. W. Hochachk & P. W. Mommsen (Eds.), Biochemistry and Molecular Biology of Fishes (Vol. 2, pp. 1–38). Elsevier, Amsterdam. [ Links ]
Montecino V. & C.B. Lange. 2009. The Humboldt Current System: Ecosystem components and processes, fisheries, and sediment studies. Progress in Oceanography, 83(1–4), 65–79. http://doi.org/10.1016/j.pocean.2009.07.041 [ Links ]
Nei M. 1973. Analysis of gene diversity in subdivided populations. PNAS, Proceedings of the National Academy ofSciences, 70(12), 3321–3323. http://doi.org/10.1073/pnas.70.12.3321 [ Links ]
NOAA (National Oceanic and Atmospheric Administration). 2015. Fishwatch: Pacific Mackerel. http://www.fishwatch.gov/profiles/pacific-mackerel [ Links ]
Ñiquen M. & M. Bouchon. 2004. Impact of El Niño events on pelagic fisheries in Peruvian waters. Deep-Sea Research Part II: Topical Studies in Oceanography, 51(6–9), 563–574. http://doi.org/10.1016/j.dsr2.2004.03.001 [ Links ]
Palumbi S.R. 1994. Genetic divergence, reproductive isolation,and marine speciation. Annual Review of Ecology andSystematics, 547–572. http://doi.org/10.1146/annurev.es.25.110194.002555 [ Links ]
Posada D. 2008. jModelTest: Phylogenetic Model Averaging. Molecular Biology and Evolution, 25(7), 1253–1256. http://doi.org/10.1093/molbev/msn083 [ Links ]
Rambaut A. & A.J. Drummond. 2009. Tracer v1.5. http://tree.bio.ed.ac.uk/software/tracer/ [ Links ]
Ramos-Onsins S.E. & J. Rozas. 2002. Statistical properties of new neutrality tests against population growth. MolecularBiology and Evolution, 19(12), 2092–2100. http://doi.org/10.1093/molbev/msl052 [ Links ]
Raymond M. & F. Rousset. 1995. An exact test for populationdifferentiation. Evolution, 49(6), 1280–1283. http://doi.org/10.2307/2410454 [ Links ]
Rein B., A. Lückge, L. Reinhardt, et al. 2005. El Niño variability offPeru during the last 20,000 years. Paleoceanography, 20, 1–18. http://doi.org/10.1029/2004PA001099 [ Links ]
Rice W.R. 1989. Analyzing Tables of Statistical Tests. Evolution, 43(1), 223. https://doi.org/10.2307/2409177 [ Links ]
Rodrigues R., H. Schneider, S. Santos, et al. 2008. Low levels of geneticdiversity depicted from mitochondrial DNA sequences in a heavily exploited marine fish (Cynoscion acoupa, Sciaenidae)from the Northern coast of Brazil. Genetics and Molecular Biology, 31(2), 487–492. http://doi.org/10.1590/S141547572008000300015 [ Links ]
Rogers A.R. & H. Harpending. 1992. Population growth makes wavesin the distribution of pairwise genetic differences. MolecularBiology and Evolution, 9(3), 552–569. [ Links ]
Salvatteci R. 2013. Variabilité multi-décennale et millénaire de l’intensité de la Zone de Minimum d’Oxygène, de la production exportée et des populations de poissons pélagiques à partir de sédiments marins laminés à Pisco, Pérou au coursdes derniers 25 000 ans. Thèse de Doctorat De L’université Pierre Et Marie Curie.
Sanchez G. & R. Calienes & S. Zuta. 2000. The 1997-98 El Niño and its effects on the coastal marine ecosystem off Peru. CalcofiReports, 41, 62–86. [ Links ]
Sanchez G., S. Tomano, C. Yamashiro, et al. 2016. Population geneticsof the jumbo squid Dosidicus gigas (Cephalopoda: Ommastrephidae) in the northern Humboldt Current system based on mitochondrial and microsatellite DNA markers. Fisheries Research, 175, 1–9. http://doi.org/10.1016/j.fishres.2015.11.005 [ Links ]
Santander H. & R. Flores. 1983. Los desoves y distribución larval de cuatro especies pelágicas y sus relaciones con las variaciones del ambiente marino frente al Perú. FAO Fish. Rept. [ Links ]
Serra J.R. 1982. Scomber japonicus peruanus. In Estado actual de las Principales Pesquerías Nacionales. Bases para un Desarrollo Pesquero. Corporación de Fomento de la Producción/Instituto de Fomento Pesquero. [ Links ]
Teacher A.G.F. & D.J. Griffiths. 2011. HapStar: Automated haplotype network layout and visualization. Molecular Ecology Resources, 11(1), 151–153. http://doi.org/10.1111/j.17550998.2010.02890.x [ Links ]
Theisen T.C., B.W. Bowen, W. Lanier, et al. 2008. High connectivity on a global scale in the pelagic wahoo, Acanthocybiumsolandri (tuna family Scombridae). Molecular Ecology,17(19), 4233–4247. http://doi.org/10.1111/j.1365294X.2008.03913.x [ Links ]
Tzeng T. 2007. Population structure and historical demography of thespotted mackerel (Scomber australasicus) off Taiwan inferred from mitochondrial control region sequencing. Zoological Studies, 46(6), 656–663. [ Links ]
Tzeng T., H. Haung, D. Wang & S. Yeh. 2007. Genetic Diversity and Population Expansion of the Common Mackerel (Scomber japonicus) off Taiwan. Journal of the Fisheries Society of Taiwan, 34(3), 237–245. [ Links ]
Utter F.M. 1994. Perspectives of molecular genetics and fisheries into the 21st century. Reviews in Fish Biology and Fisheries, 4(3),374–378. http://doi.org/10.1007/BF00042910 [ Links ]
Viñas J., J.R. Alvarado Bremer & C. Pla. 2004. Inter-oceanic genetic differentiation among albacore (Thunnus alalunga) populations. Marine Biology, 145(2), 225–232. http://doi.org/10.1007/s00227-004-1319-5 [ Links ]
Wang M.L., X.M. Zhang, T.Y. Yang, et al. 2008. Genetic diversity in the mtDNA control region and population structure in the Sardinella zunasi Bleeker. African Journal Biotechnology, 7(24), 4384–4392. [ Links ]
Ward R.D. 2000. Genetics in fisheries management. Hydrobiologia, 420(1–3), 191–201. http://doi.org/10.1023/A:1003928327503 [ Links ]
Ward R.D., M. Woodwark & D.O.F. Skibinski. 1994. A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. Journal of Fish Biology, 44(2), 213–232. http://doi.org/10.1111/j.1095-8649.1994.tb01200.x [ Links ]
Waters J. & M.S. Roy. 2004. Phylogeography of a high-dispersal New Zealand sea-star: does upwelling block gene-flow? MolecularEcology, 13(9), 2797–806. http://doi.org/10.1111/j.1365294X.2004.02282.x [ Links ]
Yagishita, N. & T. Kobayashi. 2008. Isolation and characterization of nine microsatellite loci from the chub mackerel, Scomber japonicus (Perciformes, Scombridae). Molecular EcologyResources, 8(2), 302–304. https://doi.org/10.1111/j.14718286.2007.01935.x [ Links ]
Yan S., G. Catanese, C.L. Brown, et al. 2015. Phylogeographic study onthe chub mackerel (Scomber japonicus) in the NorthwesternPacific indicates the late Pleistocene population isolation. Marine Ecology, 36(3), 753–765. http://doi.org/10.1111/maec.12267 [ Links ]
Zardoya R., R. Castilho, C. Grande, et al. 2004. Differential population structuring of two closely related fish species, themackerel (Scomber scombrus) and the chub mackerel(Scomber japonicus), in the Mediterranean Sea. Molecular Ecology, 13, 1785–1798. http://doi.org/10.1111/j.1365294X.2004.02198.x [ Links ]
Zeng L., Q. Cheng & X. Chen. 2012. Microsatellite analysis reveals the population structure and migration patterns of Scomber japonicus (Scombridae) with continuous distributionin the East and South China Seas. Biochemical Systematics and Ecology, 42, 83–93. http://doi.org/10.1016/j.bse.2012.02.014 [ Links ]
Zeng, L. & Q. Cheng. 2012. Thirty novel microsatellite markers for the coastal pelagic fish, Scomber japonicus (Scombridae). Journal of Genetics, supl. Supplement; London 91, 1–5. https://doi.org/10.1007/s12041-012-0153-7 [ Links ]
Zhu Y., Q. Cheng & S.M. Roger. 2014. Genetic structure of Scomber japonicus (Perciformes: Scombridae) along the coast ofChina revealed by complete mitochondrial cytochrome b sequences. Mitochondrial DNA. Part A, DNA Mapping, Sequencing, and Analysis, 1–9. http://doi.org/10.3109/19401736.2014.958671 [ Links ]
Información sobre los autores:
SB y DO diseñaron el estudio. SB realizó el trabajo de colecta. DO asesoró con el análisis de los datos y la metodología en laboratorio. SB redactó el trabajo. DO y RQ contribuyeron con la discusión del trabajo. SB, DO y RQ revisaron y aprobaron el manuscrito.
Los autores no incurren en conflictos de intereses.
Fuentes de financiamiento: Universidad Nacional Mayor de San Marcos, Vicerrectorado de Investigación, Proyecto Nº 141001027.
Presentado: 01/12/2016
Aceptado: 08/06/2017
Publicado online: 20/07/2017
