










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa Médica Peruana
versión On-line ISSN 1728-5917
Acta méd. peruana v.23 n.2 Lima mayo/agos. 2006
SIMPOSIO: HIPERTENSIÓN ARTERIAL
Fisiología de la Hipertensión Arterial esencial
Physiopathology of essential arterial hypertension
Raúl Gamboa A1
1 Profesor Emérito e Investigador de la Universidad Peruana Cayetano Heredia. Médico Cardiólogo
RESUMEN
La hipertensión arterial es altamente prevalente en la sociedad moderna y no obstante los notables avances en el conocimiento de sus mecanismos, el impacto sobre su control es universalmente pobre. Sin embargo, es tal la complejidad de los mecanismos interactuantes, que por lo general el método más frecuentemente utilizado para su tratamiento es el del ensayo / error considerando variables fenotípicas resultantes de la interacción ambiente/genotipo y respetando las condiciones básicas de: a) efectividad, b) sin perjudicar la calidad de vida, c) sin propiciar el desarrollo de factores de riesgo, d) sin antagonismos con la comorbilidad, y e) dirigidos a la protección de los órganos blanco. Idealmente el tratamiento del paciente hipertenso esencial, debería basarse en el conocimiento de los mecanismos fisiopatológicos que contribuyen a su desarrollo. Los avances actuales en genética, en el genoma humano, integrados a los conocimientos fisiológicos, fisiopatológicos y estudios de población permitirán en el futuro un tratamiento más racional y selectivo, y aún la prevención de la hipertensión arterial.
PALABRAS CLAVES: fisiopatología de la Hipertensión arterial, tratamiento, paciente hipertenso.
ABSTRACT
Arterial hypertension has a high prevalence in modern society and despite the great advances in our knowledge of its mechanism, the impact of its control is poor general. Although this complex mechanism interact, the method more frequently used of its treatment is the essay error consider phenotype variables as result of interaction of environmental genotype and respect of basic conditions such as a) effectivity, b) with out harming the quality of life, c) without propitiating the development of factors of risk d) without oppositions with the co-morbidity and e) directed to the protection of the organs target. Treatment ideally of essential hypertension patient should base on the physiopathologic mechanisms knowledge that contributes to its development. The current genetic and human genome advances integrates of physiologic and physiopathologic knowledge and population researches led us in the future reach a more selective and rational treatment even the arterial hypertension prevention.
KEY WORDS: Physiopathology of essntial arterial hypertension, treatment, hypertensive Patient.
El sistema circulatorio humano es una intrincada red de mecanismos destinados a mantener la homeostasis de presión y flujo pese a numerosas perturbaciones. Por tanto, una elevación constante de la presión arterial refleja un trastorno en las delicadas interrelaciones de los factores que mantienen este equilibrio. La hipertensión arterial esencial, o hipertensión de causa no determinada, es responsable de más del 90% de los casos de hipertensión vistos en la práctica médica. El hallazgo tiende a aparecer con carácter familiar más que individual y es representativo de una colección de enfermedades o síndromes, basados genéticamente en anormalidades dependientes de una interacción ambiente genotipo, y en consecuencia con diferentes severidades y tiempos de aparición(1-3).
Son muchos los factores fisiopatológicos que han sido considerados en la génesis de la hipertensión esencial: el incremento en la actividad del sistema nervioso simpático (SNS), tal vez relacionado con excesiva exposición o respuesta al estrés psicosocial de la culturaleza, es decir del impacto de la vida moderna; la sobreproducción de hormonas ahorradoras de sodio y vasoconstrictoras; la alta ingesta de sodio; la inadecuada ingesta de potasio y calcio; el incremento en la secreción o la inapropiada actividad de la renina, con el resultante incremento en la producción de angiotensina II y aldosterona (SRAA); la deficiencia de vasodilatadores, tales como la prostaciclina, el óxido nítrico (ON) y los péptidos natriuréticos; la alteración en la expresión del sistema kinina-kalikreína, que afecta el tono vascular y el manejo renal del sodio; las anormalidades en los vasos de resistencia, incluyendo lesiones en la microvasculatura renal; la diabetes mellitus, la resistencia a la insulina; la obesidad; el incremento en la actividad de factores de crecimiento; las alteraciones en los receptores adrenérgicos, que influencian la frecuencia cardiaca, el inotropismo cardiaco y el tono vascular; y las alteraciones celulares en el transporte iónico(4).
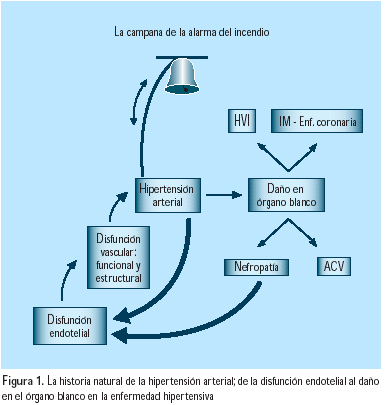
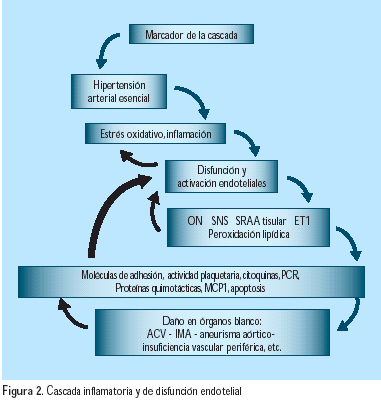
El nuevo concepto de que las anormalidades funcionales y estructurales, incluyendo la disfunción endotelial, el incremento del estrés oxidativo, la remodelación vascular y la reducción de la complacencia, pueden anteceder a la hipertensión y contribuir a su patogénesis ha ganado soporte en los últimos años; parece evidente que la hipertensión arterial sería tal vez la campana de alarma del síndrome(5) (Figura 1) y el inicio de una verdadera cascada, siguiendo a la inflamación y disfunción endotelial (Figura 2). Aunque son diversos los factores que contribuyen a la patogénesis del mantenimiento de la elevación de la presión arterial, los mecanismos renales probablemente juegan un rol primario, tal como fuera planteado por Guyton(6), en 1991, al decir que la presión arterial empieza a elevarse cuando los riñones requieren de mayor presión que la usual, para mantener el volumen de los líquidos extracelulares dentro de los límites normales.
GENÉTICA
Las evidencias de influencias genéticas en el desarrollo de la hipertensión esencial provienen de diferentes fuentes. Las investigaciones en hermanos gemelos documentan mayor concordancia de presiones arteriales entre los monocigotos que entre los dizigotos(7,8). Hoy se acepta que la hipertensión arterial esencial es mayormente un síndrome con compromiso multifactorial y generalmente poligénico y familiar. Menos del 5% de los hipertensos tiene una causa monogénica de mecanismo mendeliano, es decir con transmisión de rasgos codificados por un solo gen. Entre estas raras formas de hipertensión arterial, la mayoría relacionadas con la regulación renal del sodio y manejo del agua, figuran: a) la hipertensión glicocorticoide remediable, manifestada por la sobreproducción de aldosterona y actividad mineralocorticoide incrementada; b) el exceso aparente de mineralocorticoides, caracterizado por altos niveles de cortisol; c) el síndrome de Liddle, expresado por un incremento en la reabsorción del sodio; d) el seudohermafroditismo masculino y femenino; e) el síndrome de Gordon, originado por un defecto renal en el transporte iónico; f) la hipertensión y bradilactilia; g) la hipertensión debida a feocromocitoma; y, h) los más prometedores hallazgos se relacionan con los genes del SRAA, tal como la variante M235T en el gen de angiotensinógeno, el cual ha sido asociado con incrementos en los niveles de angiotensinógeno circulante y presión arterial(9-12). Sin embargo, estas alteraciones monogénicas se correlacionan solo con modestos cambios en la presión arterial. Observaciones en ingeniería genética en ratones han demostrado la posibilidad de generar modelos transgénicos hipertensivos(13).
Las diversas formas monogénicas de hipertensión arterial pueden manifestar gran variabilidad fenotípica. Aunque la causa de las hipertensiones monogénicas han sido atribuidas al defecto de un gen individual, es común observar que indivíduos con el mismo defecto genético varían en la severidad de la enfermedad; este fenómeno se le conoce como penetrancia variable(14).
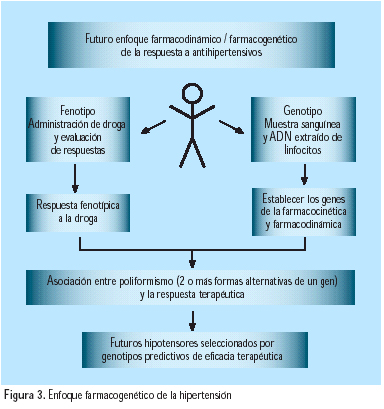
Se estima que entre las familias hipertensas, 30 a 60% de ellas tienen una base genética poligénica. La respuesta hipertensiva constituye una respuesta fenotípica a la interacción entre el factor o factores ambientales y el genotipo (Figura 4). El polimorfismo fisiopatológico y genético de la hipertensión esencial la alejan de la monoterapia. Un gran aporte futuro de la genética se enfocará en la farmacogenética, es decir en la posibilidad de seleccionar futuras drogas hipotensoras en base al conocimiento de genotipos predictivos de eficacia terapéutica(15), tal y como se describe en la Figura 3.
ASOCIACIÓN HEREDITARIA ENTRE FACTORES DE RIESGO CARDIOVASCULAR
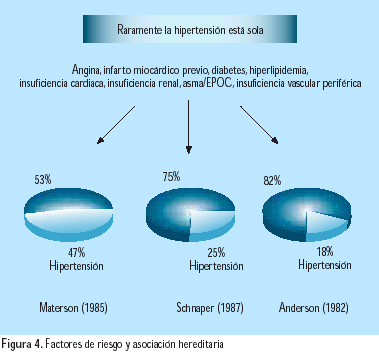
Los factores de riesgo, incluyendo la hipertensión arterial, tienden frecuentemente a agregarse(7,8) (Figura 4). Aproximadamente, 40% de las personas con hipertensión arterial es también hipercolesterolémica. Estudios genéticos han establecido una clara asociación entre hipertensión y dislipidemia(16). La hipertensión y la diabetes mellitus tipo 2 también coexisten. La hipertensión es dos veces más frecuente en personas con diabetes que en aquellas sin diabetes(17). La mayor causa de mortalidad en pacientes con diabetes tipo 2 es la enfermedad coronaria, y la diabetes incrementa el riesgo de infarto miocárdico agudo (IMA), tanto como un previo IMA en una persona no diabética(17). Desde que de 35% a 75% de las complicaciones cardiovasculares de la diabetes son atribuibles a la hipertensión, los pacientes diabéticos requieren un agresivo manejo antihipertensivo, así como un adecuado tratamiento y control de la dislipidemia y de la glicemia. La asociación de hipertensión, resistencia a la insulina, dislipidemia, obesidad, microalbuminuria e hiperuricemia, con frecuencia se asocian en el denominado síndrome metabólico, asociación con gran impacto en el riesgo de enfermedad cardiovascular(18).
SISTEMA NERVIOSO SIMPÁTICO
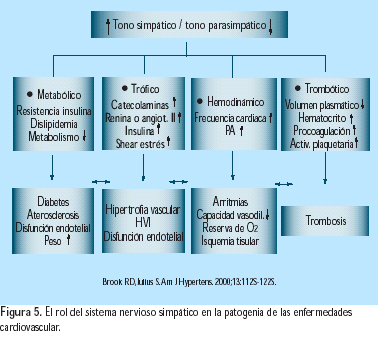
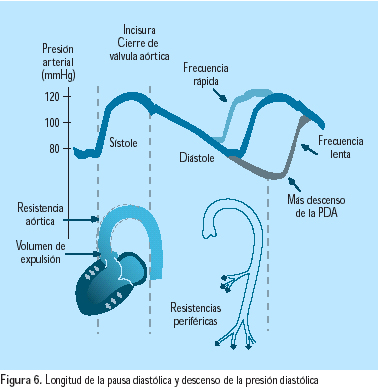
El incremento en la actividad del SNS incrementa la presión sanguínea y contribuye al desarrollo y mantenimiento de la hipertensión a través de la estimulación del corazón, vasculatura periférica y riñones, causando incremento en el gasto cardiaco, en la resistencia vascular y en la retención de líquidos(19). Además, el desbalance autonómico (incremento del tono simpático y reducción del tono parasimpático) ha sido asociado con anormalidades metabólicas, hemodinámicas, tróficas y reológicas, resultantes en incrementos en morbilidad y mortalidad cardiovascular(20) (Figura 5). Diversos estudios han demostrado la relación entre la frecuencia cardíaca y el desarrollo de hipertensión diastólica. Es conocida la relación entre la longitud de la pausa diastólica y el descenso de la presión diastólica(21) (Figura 6). Las evidencias indican que los incrementos en la frecuencia cardíaca son originados mayormente por reducción en el tono parasimpático, soportando así el concepto de que el desbalance autonómico contribuye a la patogénesis de la hipertensión arterial. Además, desde que el nivel de la presión diastólica se relaciona más cercanamente a la resistencia vascular que a la función cardíaca, es sugestivo que el incremento del tono simpático puede también incrementar la presión diastólica, al causar proliferación de las células vasculares lisas y en consecuencia remodelación vascular. El incremento de la estimulación simpática es mayor en los jóvenes, lo cual puede contribuir significativamente al desarrollo de la hipertensión en edades tempranas(22). Los mecanismos del incremento de la actividad simpática son complejos e involucran alteraciones en baro y quimorreceptores. Los barorreceptores arteriales son reajustados a nivel más alto en los pacientes hipertensos, principalmente por acción de la angiotensina II y por el efecto de radicales libres y endotelina(23). La exagerada respuesta a quimiorreceptores, que conduce a incremento en la actividad simpática, ha sido demostrada con estímulos tales como el apnea y la hipoxia(24). La crónica estimulación simpática conduce a remodelación vascular y a hipertrofia ventricular izquierda, posiblemente por el efecto directo de la epinefrina en sus receptores, así como por la liberación de factores tróficos, tales como el factor de crecimiento ß transformante, el factor 1 de crecimiento semejante a la insulina y el factor de crecimiento fibroblástico(20). La estimulación simpática renal también está incrementada en los pacientes hipertensos. En modelos animales, la estimulación renal simpática induce reabsorción tubular de sodio y agua, así como la reducción urinaria de la excreción de sodio y agua, resultando en la expansión del volumen intravascular y el incremento de la presión arterial(25).
REACTIVIDAD VASCULAR
Los pacientes hipertensos presentan mayor respuesta vasoconstrictora a la infusión de norepinefrina que los controles normotensos(26). La mayor respuesta vasoconstrictora a la norepinefrina se observa también en los hijos normotensos de padres hipertensos añejos, comparados con controles sin historia familiar de hipertensión, sugiriendo que la hipersensitividad a la norepinefrina puede ser de origen genético y no consecuencia de la hipertensión misma.
Los agentes simpaticolíticos de acción central y los antagonistas α y ß adrenérgicos son muy efectivos en la reducción de la hipertensión arterial esencial, evidenciando la importancia de los mecanismos simpáticos en su mantenimiento. La exposición al estrés incrementa la actividad simpática y su repetida activación induce vasoconstricción arteriolar, lo que origina hipertrofia vascular y en consecuencia progresivo incremento en resistencia vascular periférica y presión arterial(27).
REMODELAMIENTO VASCULAR Y ENDURECIMIENTO ARTERIAL
La resistencia vascular periférica está característicamente elevada en la hipertensión arterial, debido a alteraciones estructurales y funcionales en las pequeñas arterias. La remodelación de estos vasos contribuye al desarrollo de la hipertensión y su asociado daño en los órganos blanco(28). La resistencia periférica se incrementa a nivel precapilar, incluyendo las arteriolas (arterias conteniendo solo una capa de células musculares lisas) y la pequeñas arterias (diámetro de luz < 300 μm). La elevada resistencia periférica en los pacientes hipertensos está relacionada con una disminución en el número de vasos y disminución de su luz, sin incrementar el grosor de la pared (remodelación eutrófica(28).
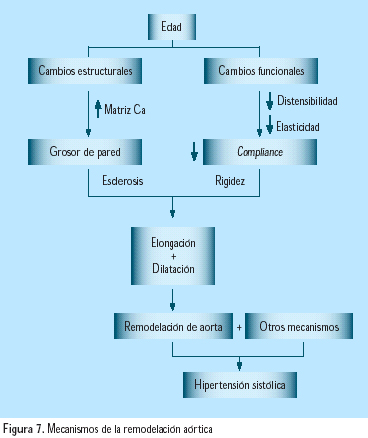
La presión sistólica y la presión del pulso se incrementan con la edad, debido principalmente a pérdida de elasticidad en las grandes arterias. La arterioesclerosis en estas arterias resulta en calcificación, depósitos de colágeno, hipertrofia de células musculares lisas, así como fragmentación de fibras elásticas en la capa media. Además de estas alteraciones estructurales, se acompaña de alteraciones funcionales debidas a la reducción en la síntesis de óxido nítrico (ON), por menor actividad de la sintasa del ON, tal vez en relación con la pérdida de función endotelial. El endurecimiento arterial contribuye a la ampliación de la presión diferencial o presión del pulso en los ancianos(29) (Figura 7).
ÁCIDO ÚRICO
La hiperuricemia está claramente asociada a hipertensión arterial y a enfermedad cardiovascular. La hiperuricemia se asocia con vasoconstricción renal(30) y se correlaciona positivamente con la actividad de la renina plasmática. Más aun, cuando ella ocurre como complicación del uso de diuréticos, se le considera como un factor de riesgo para eventos cardiovasculares(31,32), posiblemente como causante de efectos nefrotóxicos e hipertensivos.
ANGIOTENSINA II Y ESTRÉS OXIDATIVO
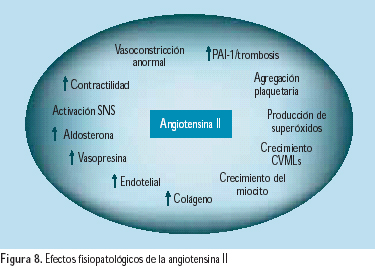
El conocimiento de los múltiples efectos fisiopatológicos del exceso de actividad del SRA y su producto final, la angiotensina II, ha conducido a la hipótesis de que los inhibidores de la enzima conversora de angiotensina (IECA) y los bloqueadores del receptor de angiotensina II (BRAs) tienen importantes efectos vasoprotectores, que van mas allá de la reducción de la presión arterial. Importantes estudios clínicos respaldan esta hipótesis(33) (Figura 8). La presencia de hipertensión arterial crea un círculo vicioso de retroalimentación, donde la hipertensión activa al sistema y este produce mayor hipertensión(34) (Figura 9).
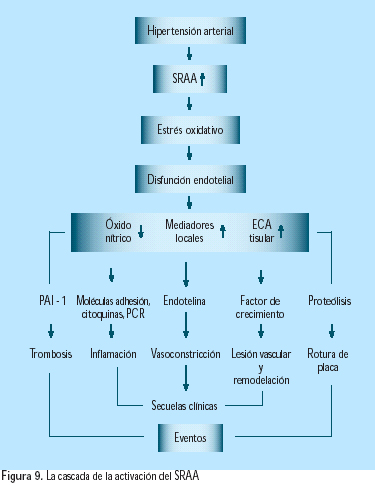
ALDOSTERONA
Los mineralocordicoides son esteroides que actúan en el epitelio renal y en otros epitelios, incrementando la reabsorción del sodio y la excreción del potasio e iones hidrógeno. El agua es retenida junto con el sodio, causando la expansión del volumen extracelular. Los mineralocorticoides también actúan en el cerebro, influenciando los niveles de presión arterial. La aldosterona es el mineralocorticoide más importante, teniendo acciones autocrinas y paracrinas en el corazón y en la vasculatura, estimulando la fibrosis intra y perivascular, además de la fibrosis intersticial en el corazón. Tanto el antagonista no selectivo de aldosterona, la espironolactona y el recientemente descubierto bloqueador del receptor de aldosterona, eplerenone, son efectivos en la prevención o reversión de los depósitos de colágeno, tanto en la vasculatura como en el corazón, demostrando su efecto antifibrótico. La espironolactona y el mejor tolerado eplerenone son actualmente usados para el tratamiento de la hipertensión arterial, insuficiencia cardíaca o infarto miocárdico complicado con disfunción ventricular, debido a sus efectos protectores tisulares(35).
ENDOTELINA
El estrés de flujo, la hipoxia, las catecolaminas y la angiotensina II estimulan la producción vascular de las endotelinas. La endotelina-1 ejerce un amplio rango de efectos biológicos renales, incluyendo contracción de la vasculatura, contracción del mesangio, inhibición de la reabsorción de sodio y agua por el nefrón; además, al estimular la glándula adrenal estimula la secreción de aldosterona, produciendo vasoconstricción de la arteriola aferente renal, propiciando la hipertensión intraglomerular. La endotelina-1 estimula la actividad simpática y en consecuencia la vasoconstricción arterial. En base a estas consideraciones, se postula que la endotelina participa en mecanismos que conducen a la hipertensión arterial, principalmente en pacientes con enfermedad renal crónica(36).
ENFERMEDAD RENAL MICROVASCULAR
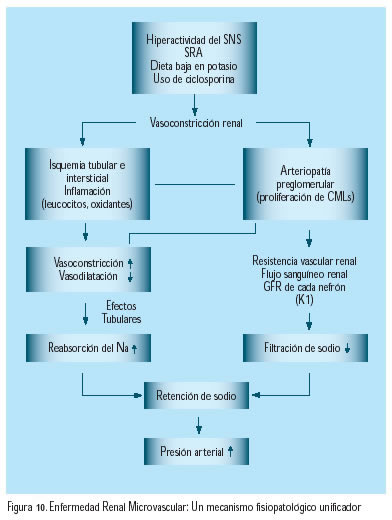
La probabilidad inicialmente propuesta por Guyton, en 1991, en relación con el rol preponderante renal en el desarrollo de la hipertensión arterial6 ha sido recientemente revivida por Johnson y colaboradores(37). Estos autores sugieren que el desarrollo del fenómeno hemodinámico general, la hipertensión arterial, se inicia con injurias renales subclínicas que conducen al desarrollo selectivo de una arteriolopatía aferente y enfermedad túbulointersticial. Los factores precipitantes serían la hiperactividad del sistema nervioso simpático y/o el incremento de la actividad del SRAA(33,34), y que el inicio de esa vía puede ser facilitada por factores genéticos que estimulan la reabsorción del sodio o limitan su filtración. Estos factores resultan en vasoconstricción renal, la cual puede conducir a isquemia renal, entrada de leucocitos y generación local de especies reactivas de oxígeno. La injuria renal estimula la generación local de angiotensina II, la cual da lugar al desarrollo de enfermedad renal microvascular, con efectos hemodinámicos glomerulares manifiestos al incrementar la resistencia arteriolar eferente, además de reducir el coeficiente de ultrafiltración y reducir la filtración del sodio, lo cual conduce a hipertensión arterial(38) (Figura 10).
MECANISMOS DE INTEGRACIÓN
El centro vasomotor bulbar es tal como los termostatos reguladores de la temperatura, es el presostato en la regulación de la presión arterial. La presión arterial es permanentemente regulada de acuerdo con lo programado en el presostato bulbar, concordante con características genéticas, ambientales y requerimientos de perfusión tisular. Cuando la homeostasis tisular requiere de mayor presión, el presostato activa al sistema nervioso simpático y este llama en su auxilio al SRAA, un sistema interactuante con el SNS. El SRAA inhibe mecanismos vasodilatadores, tales como los sistemas de kininas y péptidos natriuréticos, y activa al sistema vasoconstrictor de la endotelina. Tanto la norepinefrina como la angiotensina II abren canales de calcio a nivel vascular y cardiaco, e incrementan las resistencias periféricas y el gasto cardiaco. dando lugar al incremento de la presión arterial. La elevación crónica de la presión arterial es causa de injuria endotelial con distribución difusa, originando la reducción de los factores de relajación e incrementando el accionar de los factores de contracción derivados del endotelio. Estas respuestas son moduladas por la genética individual. La injuria endotelial compromete al endotelio glomerular, dando lugar a la glomeruloesclerosis y perturbando el balance de sodio y fluidos, lo cual contribuye al incremento de resistencias y gasto cardiaco; acompaña a estas alteraciones la aparición de la microalbuminuria, heraldo de la disfunción endotelial. La injuria endotelial da lugar a mitógenos causantes de hipertrofia vascular y fibrosis, incrementando así las resitencias periféricas. De otro lado, la injuria endotelial y la fibrosis vascular causan pérdida de sensibilidad y reprogramación de los presoreceptores y quimoreceptores, trasmitiendo así errónea información al centro vasomotor bulbar, el cual falla entonces en su rol regulador de la presión arterial. Esta secuencia fisiopatológica es descrita en la Figura 1339,(39-40).
REFERENCIAS
1. Kannel WB, Garrison RJ, Dannenberg AL. Secular trends in blood pressure in normotensive persons: the Framingham Study. Am Heart J. 1993;125:1154-8. [ Links ]
2. Wilson PWF, Kannel WB. Hypertension, other risk factors and risk of cardiovascular disease. En: Brenner BM. Hypertension: Pathophysiology. Diagnosis and Management, 2nd ed. N.Y.: Raven Press. 1995:99-114. [ Links ]
3. Carretero OA, Oparil S. Essential hypertension. Part I: definition and etiology: Circulation. 2000;101:329-35. [ Links ]
4. Calhoun DA,Bakir SE, Oparil S. Etiology and pathogenesis of essential hypertension. En: Crawford MH, DiMarco JP, eds. Cardiology. London: Mosby International. 2003;3.1-3.10. [ Links ]
5. Weber MJ. Natural history of hypertension. J Hypertension. 2003;21(suppl 6):S37-S46. [ Links ]
6. Guyton AC. Blood pressure control-special role of the kidney and body fluids. Science. 1991;252:1813-6. [ Links ]
7. Longini IM Jr. Higgins MW, Hinton PC, Moll PP, Keller JB. Environmental and genetic sources of familial aggregation of blood pressure in Tecumseh, Michigan. Am J Epidemiol. 1984;1290:131-44. [ Links ]
8. Biron P, Mongeau JG, Bertrand D. Familial aggregation of blood pressure in 558 adopted children. Can Med Assoc J. 1976;115:773-4. [ Links ]
9. Wilson FH, Disse-Nicodéme S,Choate KA, et al. Human hypertension caused by mutatios in WNK kinases. Science. 2001;293:1107-12. [ Links ]
10. Levy S, DeStefano AL, Larson Mg, et al. Evidence for a gene influencing blood pressure on chromosome 17. Framingham heart study. Hypertension. 2000;36:477-83. [ Links ]
11. Junemaitre X, Soubrier F, Kotelevtsev YV, et al. Molecular basis of human hypertension: role of angiotensinogen. Cell. 1992;71:169-80. [ Links ]
12. Staessen JA, Kuznetsova T, WangJG, et. al. M235T angiotensinogen gene polymorphism and cardiovascular renal risk. J Hypertens. 1999;17:9-17. [ Links ]
13. Mancia G. Manual of Hypertension. 2nd ed. London: Ed. Churchill Livingstone, 2002. [ Links ]
14. Luft FC. Molecular genetics of human hypertension. Curr Opin Nephrol Hypertens. 2000;9:258-66. [ Links ]
15. Cadman PE, OConnor DT. Pharmacogenomics of hypertension. Curr Opin Nephrol Hypertens. 2003;12:61-70 [ Links ]
16. Hunt SB, Ellison RC, Arwood LD, et al. Genome scans for blood pressure and hypertension. Hypertension. 2002;40:1-6. [ Links ]
17. Haffner SM, Lehto, S, Ronnemaa T, et al. Mortality from coronary heart disease in subjects with type 2 diabetes and innondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229-34. [ Links ]
18. Reaven GM. Role of insulin resistance in human disease. Diabetes. 1988;37:1595-607. [ Links ]
19. Mark AL. The sympathetic nervous system in hypertension: a potential long-term regulator of arterial pressure. J Hypertens Suppl. 1996;14:S159-65. [ Links ]
20. BrookRD, JuliusS. Autonomic imbalance, hypertension, and cardiovascular risk. Am J Hypertens. 2000;13:112S-122S. [ Links ]
21. Opie LH. The Heart. Physiology from Cell to Circulation. 1998. Philadelphia – New York: Lippincott-Raven. [ Links ]
22. Esler M. The sympathetic system and hypertension. Am J Hypertens. 2000;13:99S-105S. [ Links ]
23. Chapleau MW, Hajduczok G, Abboud FM. Mechanisms of resetting of arterial baroreceptors: an overview. Am J Med Sci. 1988;295:327-34. [ Links ]
24. Somers VK, Mark AL, Dyken ME, ClaryMP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. 1995;96:1897-904. [ Links ]
25. DiBona GF, Kopp UC. Neural control of renal function: role in human hypertension. En: Laragh JH, Breener BM, eds. Hypertension. 2nd Edition. NY: Raven Pr. 1995:1349-58. [ Links ]
26. Ziegler MG, Mills P, Dimsdale JE. Hypertensives pressor response to nor-epinephrine. Am j Hypertens. 1991;4:586-91. [ Links ]
27. Light KC. Environmental and psychosocial stress in hypertension onset and progression. En: Oparil S, Weber MA, eds. Hypertension: A Companion to Brenner and Rectors The Kidney. Philadelphia; WB Saunders. 2000:59-70. [ Links ]
28. Mulvany MJ, Alkjaer C. Small artery structure in hypertension: dual process of remodeling and growth. Hypertension. 1993;21:391-7. [ Links ]
29. Anderson GH Jr, Blakeman N, Streeten DH. The effect of age on prevalence of secondary forms of hypertension in 4429 consecutively referred patients. J Hypertens. 1994;12(5):609-15. [ Links ]
30. Breckenridge A. Hypertension and hyperuricemia. Lancet.1966;1:15-8. [ Links ]
31. SHEP Cooperative Research Group. JAMA. 2000;284:465-71. [ Links ]
32. Kjeldsen SE, Dahlof B, Devereux RB, et al. LIFE. JAMA. 2002;288:1491-8. [ Links ]
33. Oparil S, Zaman A, Calhoun DA. Pathogenesis of hypertension. Ann Intern Med. 2003;139:761-76. [ Links ]
34. Burnier M, Brunner HR. Pathophysiologic effects of Angiotensin II. Lancet. 2000;355:637-45. [ Links ]
35. Epstein M. Aldosterone and the hypertensive kidney. J Hypertension. 2001;19:829-42. [ Links ]
36. Krum H, Viskoper RJ, Lacourciere Y, et al. The effect of an endothelin-receptor antagonist, bosentan, on blood pressure in patients with essential hypertension. N Engl J Med. 1998;338:784-90. [ Links ]
37. Johnson RJ, Herrera-Acosta J, Schreiner GF, et al. Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. N Engl J Med. 2002;346:913-23. [ Links ]
38. Swales JD. The rennin-angiotensin system in essential hypertension. London: Gowe Medical Publishing,1993. [ Links ]
39. Burton AC. Physiology and Biophysics of the Circulation. Chicago: Year Book Medical Publishers. 1965. [ Links ]
40. Sheperd JT. Pathophysiology of essential hypertension. Hypertension. 1991:18 (Suppl III):76-9. [ Links ]
