










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa Médica Peruana
versión On-line ISSN 1728-5917
Acta méd. peruana v.24 n.1 Lima ene./abr. 2007
ARTÍCULOS DE REVISIÓN
Encefalopatía hepática
Hepatic encephalopathy
Carla Bustíos Sánchez1
1. Médico Gastroenteróloga. Unidad de Hígado Hospital Nacional Edgardo Rebagliati Martins – ESSALUD Profesor de Medicina de la Universidad Ricardo Palma
RESUMEN
La encefalopatía hepática es un síndrome neuro-psiquiátrico que se observa con gran frecuencia en el paciente con cirrosis avanzada. La teoría más conocida y antigua sobre la génesis de este síndrome es la hipótesis del amonio generado en el intestino grueso, según la cual éste compuesto no sería aclarado por el sistema reticulo-endotelial hepático debido a la presencia de colaterales (shunts) y pasaría directamente al cerebro cruzando con facilidad la barrera hematoencefálica. Recientemente se ha descubierto que el amonio tiene relación cercana con la modulación de la actividad del ácido gamma-aminobutírico (GABA), un neuropéptido inhibitorio potente y al mismo tiempo con la actividad de los astrocitos. El diagnóstico es fundamentalmente clínico, siendo los exámenes auxiliares como la tomografía axial computarizada, electroencefalograma y otras sólo de carácter complementario. En este artículo se revisan los mecanismos de encefalopatía hepática, así como el diagnóstico diferencial y las limitadas opciones terapéuticas con las cuales se cuenta en la actualidad.
Palabras claves: encefalopatía hepática, amonio, ácido gamma amino butírico (GABA), astrocitos, asterixis, lactulosa, metronidazol.
ABSTRACT
Hepatic encephalopathy is a neuro-psychiatric syndrome observed with great frequency in patients with advanced cirrhosis. The oldest and most popular theory about the genesis of this syndrome is the hypothesis of the ammonia generated in the large bowel, according to which this compound is not cleared by the hepatic reticulo endothelial system due to the presence of intrahepatic shunts, passing directly to the brain, easily crossing the blood brain barrier. It has recently been discovered that ammonia has a close relationship to the modulation of gamma aminobutiric acid (GABA), a potent neuroinhibitory peptide and at the same time with astrocytic activity. The diagnosis is mainly clinical, and computerized axial tomography, electroencephalography and other methods are only a complementary. In this article the mechanisms of hepatic encephalopathy, differential diagnosis and the limited therapeutic options currently available are reviewed.
Key words: hepatic encephalopathy, ammonia, gamma aminobutiric acid (GABA), astrocytes, asterixis, lactulose, metronidazole.
DEFINICIÓN
La encefalopatía hepática (EH) se define como un complejo síndrome neuropsiquiátrico potencialmente reversible, en pacientes con disfunción hepática crónica o aguda en ausencia de otros desordenes neurológicos. Se caracteriza por un amplio rango de síntomas que van desde alteraciones mínimas de la función cerebral hasta el coma profundo1.
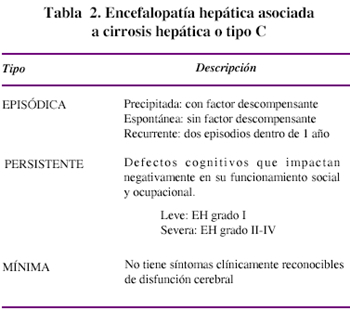
Uno de los mayores problemas que surgen, al intentar describir los diferentes aspectos clínicos de la EH, ha sido la discrepancia en las definiciones de los términos utilizados (encefalopatía portosistémica, encefalopatia aguda, coma hepático, etc), por lo cual, con el fin de usar una terminología que sea universalmente aceptada, que relacione la EH con el tipo de anormalidad hepática, las características y duración de las manifestaciones neurológicas, se la ha clasificado en tres tipos2, tal como se muestra en la Tabla 1. En esta revisión se tratará fundamentalmente sobre la EH tipo C o asociada con cirrosis hepática de acuerdo a la Tabla 2.

ETIOLOGÍA Y PATOGENIA
A pesar de ser una patología frecuente y de tener características clínicas definidas, los mecanismos etiológicos precisos involucrados en la EH no han sido totalmente establecidos. Sin embargo los estudios basados en modelos animales han identificado algunos elementos esenciales para que se produzca la EH; como son las comunicaciones portosistémicas y la alteración en la depuración hepática de metabolitos nitrogenados. Estas observaciones no explican otros parámetros potencialmente importantes como las alteraciones del metabolismo de la energía cerebral y el incremento de la permeabilidad de la barrera hematoencefálica que facilitaría el pasaje de neurotoxinas y falsos neurotransmisores al cerebro. En base a esto, un gran número de teorías han sido propuestas, de las cuales las más importantes son la teoría del amonio y del GABA.
Amonio
Es la neurotoxina mejor caracterizada que puede llevar a EH. Tradicionalmente se conoce que el amonio es producido en el tracto gastrointestinal por degradación bacteriana de aminas, aminoácidos, purinas y úrea, y que es metabolizado en el hígado, pero evidencia experimental apoya la noción de que casi todos los órganos están involucrados en el metabolismo de amonio3,4.
Normalmente el amonio es detoxificado en el hígado por conversión a úrea mediante el ciclo de Krebs y en glutamina . En la enfermedad hepática o en presencia de comunicaciones portosistémicas, el amonio sérico no es eficientemente metabolizado, incrementándose así sus niveles en sangre.
El tejido muscular también participa en el metabolismo del amonio como uno de los principales órganos que sintetizan glutamina a través de la glutamina- sintetasa. El amonio puede ser captado o liberado del músculo. El incremento del nivel de amonio en sangre ha sido observado durante el ejercicio, probablemente relacionado a la activación del ciclo nucleótido purina, y también en pacientes con enfermedad hepática crónica avanzada con poca masa muscular asociado a una menor síntesis de glutamina.
Los riñones contienen glutaminasa y glutamina sintetasa y son capaces de degradar y sintetizar la glutamina. En estados fisiológicos normales el riñón regula el ingreso del amonio a sangre y su excreción urinaria mientras que en acidosis crónica la amoniogénesis aumenta y también la excreción urinaria del amonio. A nivel del cerebro los astrocitos son el sitio de detoxi- ficación del amonio a través de la síntesis de glutamina. Actualmente se sugiere que la EH se podría producir por una disfunción primaria de estas células con una alteración neuronal secundaria3,5.
A pesar de la fuerte evidencia que implica al amonio como el factor más importante para que se produzca la EH, los mecanismos celulares precisos no son bien conocidos. Se postulan múltiples efectos neurotóxicos, que incluyen alteraciones en el transporte de aminoácidos, agua y electrolitos a través de la membrana neuronal, deterioro de la comunicación neuronal-glial e inhibición de la generación de potenciales postsinápticos inhibitorios y excitatorios6.
Los argumentos contra la hipótesis del amonio incluyen las observaciones de que aproximadamente 10% de pacientes con EH tiene niveles normales de amonio sérico; que muchos pacientes tienen elevados niveles de amonio sin evidencia de EH y además que el amonio cuando es administrado a pacientes cirróticos no induce los cambios clásicos electroencefalográficos asociados con EH4,5.
Ácido gamma-aminobutírico (GABA)
El GABA es una sustancia neuroinhibitoria producida en el tracto gastrointestinal. A nivel del sistema nervioso central el 24 a 45 % de todas las terminaciones nerviosas cerebrales son GABAérgicas. Un incremento del tono GABAérgico es observado en pacientes con cirrosis, posiblemente por la disminución del metabolismo hepático del GABA4.
Cuando el GABA cruza la barrera hematoencefálica extrapermeable en pacientes con cirrosis, interactúa con receptores postsinápticos supersensibles. Los receptores GABA, en asociación con receptores para benzodiacepinas y barbitúricos, regulan un canal selectivo del ión cloro . La unión del GABA a su receptor permite el ingreso de iones de cloro a la neurona postsináptica, llevando a la generación de un potencial postsináptico inhibitorio. La administración de benzodiazepinas y barbitúricos a pacientes con cirrosis incrementa el tono GABAérgico y predispone a depresión del estado de conciencia5.
Se ha observado además, que el incremento del amonio aumenta la síntesis y liberación en la membrana externa de la mitocondria astroglial de neuroesteroides. Algunos neuroesteroides como tetrahidroprogesterona (THP) y tetrahidrodeoxicorticosterona (THDOC), son potentes agonistas del complejo receptor GABA, en donde hay sitios específicos para los neuroesteroides, distintos a los de las benzodiazepinas y barbitúricos.
Los niveles de estos neuroesteroides se han encontrado incrementados en modelos de ratones con insuficiencia hepática y se ha observado que al inyectárseles THP o THDOC a ratones normales se indujo cambios en los astrocitos y sedación.
Cada una de estas alteraciones en el sistema de neurotransmisión GABA, potencialmente incrementa la neurotransmisión inhibitoria, contribuyendo a las manifestaciones de la EH en los pacientes con cirrosis hepática
Falsos neurotransmisores
La alteración de la concentración de catecolaminas puede jugar un rol en la patogénesis de la EH y está asociado con alteraciones en el metabolismo de los aminoácidos. En cirrosis hepática se ha encontrado niveles bajos de aminoácidos (aa) de cadena ramificada (valina, leucina e isoleucina) y elevación del nivel de aa aromáticos y triptofano en sangre y en cerebro. Altas concentraciones de fenilalanina en el cerebro pueden inhibir la tirosina 3-hidroxilasa, la enzima llave para la síntesis de neurotransmisores catecolaminérgicos. Otras aminas tales como tiramina, octopamina, y feniletanolamina son sintetizadas por vías metabólicas alternas de la tirosina compitiendo con neurotransmisores catecolaminérgicos normales, tales como la dopamina, por el mismo sitio del receptor. La depleción cerebral de dopamina y el desplazamiento de dopamina por neurotransmisores falsos puede llevar a un deterioro en la neurotransmisión dopaminérgica. La octopamina es también producida fuera del cerebro por las bacterias intestinales, y por deterioro de metabolismo hepático, su concentración en sangre puede incrementarse. Esta teoría ha estimulado numerosos estudios para definir el rol de los cambios en los patrones de aminoacidos y el deterioro de la neurotransmisión dopaminérgica, sin embargo los resultados aún son controversiales6,7.
Glutamato
Es el más importante neurotransmisor excitatorio del cerebro de los mamíferos. Como otros neurotransmisores, el glutamato está presente en varios compartimentos. Solo el glutamato del compartimento neuronal es biológicamente activo; el glutamato en el compartimento glial es un importante metabolito y está involucrado en la fijación de amonio en el cerebro. Estudios en animales con EH muestran disminución en el número y afinidad de receptores de glutamato en el cerebro, llevando a alteraciones en la neurotransmisión glutaminérgica. Estos hallazgos juntos con los cambios en la neurotransmisión GABAérgica producen alteraciones en la neurotransmisión excitatoria e inhibitoria en la EH7.
Manganeso
Incremento de los niveles del manganeso ha sido demostrado en hepatitis aguda, hepatitis crónica y desordenes congénitos como el síndrome de Alagille. Se ha reportado alto contenido de manganeso en el globus pallidus de animales, así como en tejido del cerebro de pacientes que fallecieron por EH. Además se ha observado que mineros con exposición crónica a manganeso desarrollan encefalopatía y características extra-piramidales similares a la EH. Se postula que el manganeso deteriora el metabolismo oxidativo neuronal8. El rol del manganeso en la patogénesis de la EH y la posibilidad de su quelación como tratamiento, necesitan ser evaluados en futuros estudios.
Zinc
El zinc es un elemento esencial y funciona como antioxidante. Bajas concentraciones de zinc han sido reportadas en pacientes con cirrosis y particularmente en aquellos con EH. Pacientes con falla hepática aguda y subaguda han mostrado tener bajos niveles séricos de zinc y en animales experimentales los suplementos de zinc han llevado a reducción del amonio sérico. El déficit de zinc llevaría a alteración de neurotransmisores parecidos a GABA y norepinefrina8,9. El rol de zinc en la patogénesis de la EH aún requiere mayores estudios.
DIAGNÓSTICO
Cuadro clínico
Las características clínicas de la EH incluyen un amplio rango de síntomas neurosiquiátricos que van desde signos leves de alteración de la función cerebral hasta el coma profundo. Ninguno de estos síntomas es específico para la encefalopatía y la presencia simultánea, tanto de estos síntomas como de la enfermedad hepática crónica, no es suficiente para hacer el diagnóstico de EH. Por todo esto, se debe hacer una cuidadosa evaluación neurológica y de la función motora para excluir otras enfermedades. Se debe tener en cuenta los cambios sutiles de la vida diaria, alteración del ritmo del sueño, deterioro del estado de conciencia y de la función cognitiva. En el examen de la función motora se puede encontrar aumento del tono muscular, reducción de la velocidad de los movimientos, ataxia, deterioro postural o reflejos posturales, la presencia de movimientos anormales tales como el tremor y particularmente la asterixis10.
Gradiente clínica
El sistema de graduación clínica más utilizado para la EH es el de West Haven, el cual gradúa a la EH en cuatro grados y se basa en cambios del estado de conciencia, la función intelectual y comportamiento. Ver Tabla 3. Para estadíos III y IV se puede utilizar adicionalmente la escala de coma Glasgow2,10.

Pruebas de laboratorio
El incremento del amonio arterial se puede encontrar en cerca del 90% de pacientes con EH , sin embargo también puede estar aumentado en pacientes con cirrosis hepática sin signos de EH y sus mediciones no ayudan a evaluar la evolución de la EH ni la respuesta a la terapia11. Ninguna prueba de laboratorio por sí sola hace el diagnóstico de EH. El valor principal de las pruebas de laboratorio es su utilidad para el diagnóstico diferencial de otras encefalopatías metabólicas o la detección de causas precipitantes de la EH3,4.
Pruebas neurosicológicas
Para medir las anormalidades cognitivas en pacientes sin evidencia clínica de EH , se utilizan un gran número de pruebas neuropsicológicas que tienen como función identi- ficar selectivamente anormalidades en áreas tales como la atención y la función motora fina que son característicos de la EH mínima. Entre las pruebas más comúnmente utilizadas tenemos a la prueba de conexión numérica, la prueba de símbolos digitales, la prueba del diseño de bloques, la prueba de Posner y paradigma de Sternber.
Pruebas neurofisiológicas
En el electroencefalograma (EEG) y en los potenciales evocados los hallazgos son inespecíficos y no hacen el diagnóstico de EH. Los cambios clásicos en el EEG asociados a la EH que son las ondas de baja frecuencia y alta amplitud y las ondas trifásicas, también están presentes en otras encefalopatías. La respuesta visual evocada también demuestra un patrón clásico asociado con la EH. Sin embargo estas pruebas no son de utilidad clínica común2.
Tomografía computarizada
Evalua síntomas neurológicos agudos, y descarta otras enfermedades asociadas a confusión y coma tales como el hematoma subdural, absceso cerebral o tumores. La tomografía también puede demostrar edema cerebral3.
Resonancia magnética nuclear
Pacientes con cirrosis sin evidencia clínica de EH, exhiben anormalidades de alta señal, simétricas en el globus pallidum en las imágenes T1. La inspección profunda de las señales T1 indican un incremento en la sustancia blanca y estructuras extrapiramidales. Estas anormalidades se hacen más evidentes cuando la disfunción hepática aumenta. La acumulación de manganeso puede explicar también los hallazgos en T112.
Resonancia magnética nuclear con espectroscopia
Esta prueba con apropiado control de calidad y estandarización puede ayudar a cuantificar sustancias químicas en el cerebro. Se ha demostrado un incremento de la glutamina y disminución de la concentración del mio-inositol y colina en la EH13.
Tomografía con emisión de positrones
Esta técnica provee imágenes del cerebro que reflejan un específico proceso fisiológico o bioquímico. Ofrece la posibilidad de investigar el flujo sanguíneo cerebral, consumo de glucosa y oxígeno, disponibilidad neuro-receptora y utilización neurotransmisora12.
DIAGNÓSTICO DIFERENCIAL
Distinguir la EH de otras causas agudas y crónicas de alteración del estado mental, puede ser difícil en pacientes con cirrosis hepática. Algunas de las enfermedades que se enumeran en la Tabla 4 pueden ser diagnosticadas con la ayuda de la tomografía cerebral y pruebas séricas apropiadas.

TRATAMIENTO
El manejo de la EH se basa en mantener y minimizar complicaciones médicas del paciente con cirrosis hepática, en corregir los factores precipitantes y en actuar directamente sobre los mecanismos fisiopatológicos involucrados en la EH, tal como se detalla posteriormente. Ver Figura 1.
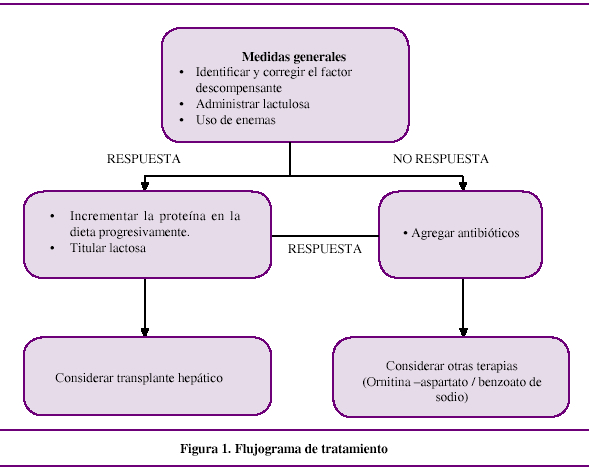
Medidas generales
Entre las medidas generales es importante monitorizar la función cardiovascular, respiratoria y renal; mantener los parámetros metabólicos (electrolitos, glucosa, etc) y manejar los problemas asociados a la enfermedad hepática crónica como la ascitis, la hemorragia digestiva alta y las infecciones virales hepatotrópicas entre otros.
Identificar y tratar los factores precipitantes
La EH en muchos pacientes es resultado de factores precipitantes. Muchas de las causas son tratables y la terapia está asociada con una rápida mejoría de la EH. Los factores precipitantes descritos en la Tabla 4, deben ser identificados y corregidos antes del diagnóstico final.
Tratamiento específico
El tratamiento específico tiene como finalidad actuar sobre las teorías involucradas en la patogénesis de la EH.
1. Disminuir el sustrato del amonio
* Proteínas de la dieta
En el pasado, se recomendaba rutinariamente la restricción de las proteínas. Sin embargo se ha observado que es más importante disminuir la producción de aminoácidos a partir de proteínas endógenas que restringir las proteínas de la dieta14. Recomendaciones:
Tan pronto se pueda reiniciar la vía oral, se debe aumentar progresivamente las proteínas hasta lograr de 1-1.5 g/kg/día o la dosis máxima tolerada.
Una dieta con proteinas de origen vegetal es mejor tolerada que con proteínas animales. Debido al mayor contenido de fibra, catártico natural, y a los bajos niveles de aminoacidos aromáticos.
*Limpieza intestinal
Se conoce que las toxinas responsables de EH se forman en el intestino y que esto es favorecido por el estreñimiento y el contenido bacteriano.
Recomendaciones:
Uso de catárticos como los disacáridos no absorbibles que producen una diarrea osmótica.
Uso de enemas: lactulosa o manitol
2. Disminuir la amoniogénesis
*Uso de disacáridos no absorvibles
La lactulosa (galactósido-fructosa), es un disacárido que no puede ser hidrolizado por las disacaridasas intestinales, y que actúa produciendo una diarrea osmótica y alterando el metabolismo de las bacterias. La lactulosa estimula la incorporación de amonio dentro de las proteínas bacterianas, reduciendo la cantidad disponible para la absorción intestinal. Además la fermentación bacteriana disminuye el pH colónico reduciendo la absorción del amonio por difusión no-iónica y promoviendo también el crecimiento de lactobacilos no productores de amonio15.
Recomendaciones:
Uso de lactulosa a dosis de 45 ml por vía oral o por sonda nasogástrica, cada hora hasta que la evacuación ocurra. Luego la dosis debe ser ajustada hasta lograr 2 a 3 deposiciones por día ( 15-45 ml cada 8-12h).
Lactulosa en enema (300 ml en 1 litro de agua) es retenido por 1 hora, con el paciente en posición de Trendelenburg.
*Uso de antibióticos
Los antibióticos se utilizan cuando no hay respuesta al tratamiento con disacáridos no absorbibles. Actúan reduciendo la formación de amonio por las bacterias.
Los antibióticos de elección son los no absorbibles, como la neomicina que actúa a nivel de las bacterias colónicas, deteriorando la actividad de la glutaminasa en las vellosidades intestinales. Puede causar nefrotoxicidad y ototoxicidad por uso prolongado o incluso precipitar un sidrome hepato-renal pese a su absorción mínima por vía oral. Otro antibiótico que puede ser utilizado es el metronidazol que actúa reduciendo la amoniogénesis, sin embargo puede asociarse a neurotoxicidad y provocar malabsorción intestinal o superinfección por estafilococos16.
Recomendaciones:
Neomicina a dosis de 3-6 g p.o por un período de 1a 2 semanas
Metronidazol a dosis de 250 mg b.i.d *Modificacion de bacterias intestinales Uso de Lactobacillus acidophilus inhibe el crecimiento de bacterias protolíticas17.
Recomendaciones:
No se ha reportado beneficios para el manejo agudo.
3. Fijación metabólica del amonio
*L-Ornitina-L-Aspartato
Es una sal estable de aminoácidos: ornitina y ácido aspártico. Su uso está basado en el concepto de que la ornitina y el aspartato son convertidos a glutamato reduciendo las concentraciones de amonio. Está disponible en formulaciones vía oral y los resultados de su uso son alentadores18,19.
*Benzoato de sodio
Fijador metabólico del amonio, por cada mol de benzoato 2 mol de nitrógeno es excretado en orina. La dosis es de 10 g al día.
*Reemplazo de zinc
El déficit de zinc es frecuente en pacientes cirróticos. La dosis utilizada es el acetato de zinc a dosis de 220 mg b.i.d.
*Alfa-ceto análogos de aminoacidos Derivados de aminoácidos deaminados, conocidos como ceto ácidos, se combina con el amonio para generar aminoácidos20.

PREVENCIÓN
La prevención de la EH debe realizarse principalmente en pacientes que se han recuperado de un episodio de EH o en aquellos a quienes se les ha colocado un shunt portosistémico intrahepático transyugular (TIPS). Dentro de las medidas para prevenir la EH se incluyen las siguientes:
Evitar los factores precipitantes
Mejorar el estado nutricional, utilizando la máxima cantidad de proteína tolerada, de preferencia de origen vegetal. Puede ser considerado, el uso de aminoácidos de cadena ramificada, en pacientes intolerantes a las proteínas,
Uso de lactulosa con la finalidad de lograr 2 a 3 movimientos intestinales/día.
Los antibióticos son reservados para pacientes quienes no responden a disacáridos no absorbibles o quienes no exhiben acidificación de heces o diarrea. Su uso crónico debe monitorizarse
Referir a transplante hepático.
AGRADECIMIENTOS
Agradecemos a la Sociedad Peruana de Medicina Interna por la autorización de reproducción parcial del contenido del libro denominado: Tópicos Selectos en Medicina Interna: Gastroenterología.
REFERENCIAS BIBLIOGRÁFICAS
1. Butterworth RF. Pathophysiology of hepatic encephalopathy: a new look at ammonia. Metab Brain Dis. 2002 ;17:221-7 [ Links ]
2. Ferenci P, Lockwood A, Mullen K, Tarter R, Weissenborn K, Blei T. Hepatic encephalopathy - Definition, nomenclature, diagnosis, and quantification: Final report of the Working Party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology 2002; 35: 716-721. [ Links ]
3. Ong JP, Aggarwal A, Krieger D, Easley KA, Karafa MT, Van Lente F, Arroliga AC, Mullen KD. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med. 2003 ;114:188-93. [ Links ]
4. Jones EA. Ammonia, the GABA neurotransmitter system, and hepatic encephalopathy . Metab Brain Dis. 2002 ;17:275-81. [ Links ]
5. Helewski K, Kowalczyk-Ziomek G, KoneckiJ. D. Ammonia and GABA-ergic neurotransmission in pathogenesis of hepatic encephalopathy Wiad Lek.2003;56:560-563. [ Links ]
6. Romero-Gomez M, Ramos-Guerrero R, Grande L, de Teran LC, Corpas R. Intestinal glutaminase activity is increased in liver cirrhosis and correlates with minimal hepatic encephalopathy. J Hepatol. 2004;41: 49-54. [ Links ]
7. Nie YQ, Zeng Z, Li YY, Sha WH, Ping L, Dai SJ. Long-term efficacy of lactulose in patients with subclinical hepatic encephalopathy. Zhonghua Nei Ke Za Zhi. 2003; 42: 261-3. [ Links ]
8. Lizardi-Cervera J, Almeda P, Guevara L, Uribe M. Hepatic encephalopathy: a review. Ann Hepatol. 2003; 2:122-130. [ Links ]
9. Vaquero J, Polson J, Chung C, Helenowski I, Schiodt FV. Infection and the progression of hepatic encephalopathy in acute liver failure.Gastroenterology 2003;125:755-764. [ Links ]
10. Als-Nielsen B, Kjaergard LL, Gluud C. Benzodiazepine receptor antagonists for acute and chronic hepatic encephalopathy. Cochrane Database Syst Rev. 2001;(4):CD002798. [ Links ]
11. Ong J, Aggarwal A, Krieger D. Correlation between ammonia level and the severity of hepatic encephalopathy. Am J Med. 2003; 114:188-193 [ Links ]
12. Nadim Joni Shah, Heiko Neeb, Maxim Zaitsev. Quantitative T1 mapping of hepatic encephalopathy using magnetic resonance imaging. Hepatology 2003; 38: 1219-1226 [ Links ]
13. Miese F; Kircheis G; Wittsack HJ, H-MR spectroscopy, magnetization transfer, and diffusion-weighted imaging in alcoholic and nonalcoholic patients with cirrhosis with hepatic encephalopathy. AJNR Am J Neuroradiol. 2006; 27:1019-1026. [ Links ]
14. Cordoba J, Lopez-Hellin J, Planas M, Sabin P, Sanpedro F, Castro F, Esteban R, Guardia J. Normal protein diet for episodic hepatic encephalopathy: results of a randomized study. J Hepatol. 2004; 41: 38-43. [ Links ]
15. Als-Nielsen B, Gluud LL, Gluud C. Nonabsorbable disaccharides for hepatic encephalopathy. Cochrane Database Syst Rev. 2004;(2):CD003044. [ Links ]
16. Maddrey W. Role of antibiotics in the management of hepatic encephalopathy. Rev Gastroenterol Disord. 2005; 5 Suppl 1:S3-9 [ Links ]
17. Boca M, Vyskocil M, Mikulecky M . Complex therapy of chronic hepatic encephalopathy completed with probiotic: comparison of two studies Cas Lek Cesk. 2004;143: 324-328. [ Links ]
18. Kircheis G, Nilius R, Held C: Therapeutic efficacy of L-ornithine-L-aspartate infusions in patients with cirrhosis and hepatic encephalopathy: results of a placebo-controlled, double-blind study. Hepatology 1997; 25: 1351-1360. [ Links ]
19. Kircheis G, Wettstein M, Dahl S, Haussinger D. Clinical efficacy of L-ornithine-L-aspartate in the management of hepatic encephalopathy. Metab Brain Dis. 2002;17:453-462. [ Links ]
20. Albrecht J, Zielinska M. The role of inhibitory amino acidergic neurotransmission in hepatic encephalopathy: a critical overview. Metab Brain Dis. 2002 ;17:283-294. [ Links ]
CORRESPONDENCIA:
Carla Bustios
