










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa Médica Peruana
versión On-line ISSN 1728-5917
Acta méd. peruana vol.32 no.4 Lima oct./dic. 2015
ARTICULO DE REVISIÓN
Gestión de riesgos en los laboratorios clínicos
Risk management in clinical laboratories
Luis Edgardo Figueroa-Montes1
1 Médico patólogo clínico. Servicio de Patología Clínica del Hospital III Suárez Angamos, EsSalud. Lima, Perú. Secretario de Acción Científica de la Asociación Médica Peruana de Patología Clínica.
RESUMEN
Las pruebas de laboratorio tienen un impacto crítico en la toma de decisiones médicas. Sin embargo, en el ciclo, que inicia con la solicitud del médico y termina con la interpretación final, pueden ocurrir errores en cualquier etapa. La evaluación de las causas que generan estos errores y la toma de medidas para detectarlos y prevenirlos, antes de que causen daño, es crítico en el proceso analítico. Esto se logra a través de la gestión de riesgos (GR). La norma EP23A, Control de calidad en el laboratorio basado en la gestión de riesgos, del Clinical Laboratory Standards Institute (CLSI), introduce los principios de la GR para los laboratorios clínicos. Esta directriz permite desarrollar un plan de GR, individualizado. Este artículo resume los principios de la GR en los laboratorios clínicos.
Palabras Clave: gestión de riesgos; control de calidad; errores médicos; laboratorio clínico
ABSTRACT
Laboratory tests have a critical impact on medical decision making. However, in the cycle, which starts with the physician’s request and ends with the final interpretation, errors may occur at any stage. The evaluation of the causes of these errors and to take measures to detect and prevent them before they cause harm, is critical in the analytical process. This is achieved through risk management (RM). EP23 Guideline: quality control based risk management laboratory, of the Clinical Laboratory Standards Institute (CLSI), introduces the principles of RM for clinical laboratories. This directive allows develop a plan of RM, individualized. This article summarizes the principles of the RM in clinical laboratories.
Key words: risk management; quality control; medical errors; clinical laboratory
INTRODUCCIÓN
El 24 de marzo de 2015, el vuelo 9525 de la aerolínea Germanwings, de Barcelona (España) hacia Düsseldorf (Alemania), operado por un avión Airbus A320-211, se estrelló en el macizo de Estrop, en los Alpes franceses de Provenza, cerca de la localidad de Barcelonnette, produciendo la muerte de 150 personas.1 Este accidente aéreo se trata de la peor catástrofe de la aviación europea en los últimos cinco años. Las autoridades alemanas, españolas y francesas y un portavoz de Germanwings anunciaron que, según las grabaciones de las cajas negras recuperadas el día anterior, la caída de la aeronave fue provocada deliberadamente. El copiloto, de 27 años de edad, estrelló el aparato de manera voluntaria, debido a su estado emocional de depresión El piloto le había cedido los mandos mientras iba al baño; cuando volvió, el copiloto no le abrió la puerta de la cabina y no contestó a las llamadas realizadas desde la torre de control. Este accidente aéreo, ¿pudo evitarse?
A pesar de que la implementación de la gestión de riesgos o gestión del riesgo (GR) nació en la industria aeronáutica militar, cada vez existen nuevas evidencias de que la GR siempre es vulnerable ante nuevos eventos. Estas vulnerabilidades en los procesos de los diferentes campos de la industria permiten establecer nuevos mecanismos, para evitar nuevos peligros y poder controlar nuevos riesgos; de esta manera, se evitarán catástrofes o fatalidades en los clientes o pacientes. Por lo tanto, la GR es un círculo que se retroalimenta y dependerá de que los gestores se involucren en su ejecución y monitorización.
El sector salud no es ajeno a esta realidad y la GR, aplicada en los establecimientos de salud, debería desarrollarse como parte de sus actividades asistenciales, a cargo de personal capacitado, para obtener excelentes resultados en su implementación y seguimiento.
Actualmente, en los laboratorios clínicos, la implementación de la GR se está desarrollando de manera paulatina, a raíz de la publicación de normas publicadas por el Clinical and Laboratory Standards Institute (CLSI), lo que obliga a conocer más sobre la GR, en beneficio de los pacientes y de la calidad analítica.
DEFINICIONES
El peligro es una fuente, situación o acto con potencial de daño, en términos de lesión y/o enfermedad. Mientras que riesgo es la combinación de la probabilidad de que ocurra un daño y la gravedad de dicho daño.2 Por eso es que muchos especialistas en la GR refieren que debería llamarse gestión de peligros. Siendo así, los peligros se identifican y los riesgos se evalúan.
La GR tiene una larga historia en otras industrias y es ampliamente utilizada por los fabricantes de dispositivos de diagnóstico in vitro. Esta GR es una forma organizada de evaluar lo que podría salir mal e identificar qué se puede hacer para mitigar el daño causado por estos errores. Identificar los peligros reales o potenciales de daño, determinar los modos de falla, priorizar su importancia y tomar acciones para mitigarlos o reducir su impacto, son la esencia de la GR.3
MAtRICES PARA EL ANÁLISIS DE LOS RIESGOS
Para realizar el análisis de los peligros y riesgos existen matrices, con escalas determinadas para su puntuación. Este análisis puede ser cualitativo o cuantitativo.
-
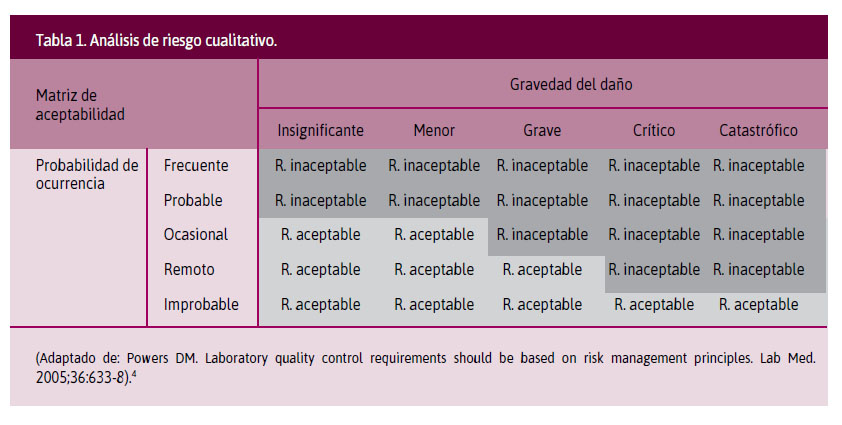
Análisis cualitativo. Se cruzan las variables probabilidad de ocurrencia (desde improbable hasta frecuente) y gravedad del daño (de insignificante a catastrófica), establecidas en una matriz de aceptabilidad, identificando riesgos aceptables o inaceptables (Tabla 1).
-
Análisis cuantitativo. Se cruzan las variables probabilidad de ocurrencia (PO), gravedad del daño (S) y se adiciona la detectabilidad (D). En este análisis se asigna una puntuación a cada variable, según la escala que utilicemos, obteniendo el producto final de multiplicar PO x S x D. Al resultado de este producto se le conoce como risk priority number (RPN o número de prioridades de riesgo). Al obtener el RPN, se puede priorizar, según los puntajes obtenidos, el riesgo que se deberá intervenir.4 En la Tabla 2 se incluyen dos ejemplos de la aplicación de esta matriz.
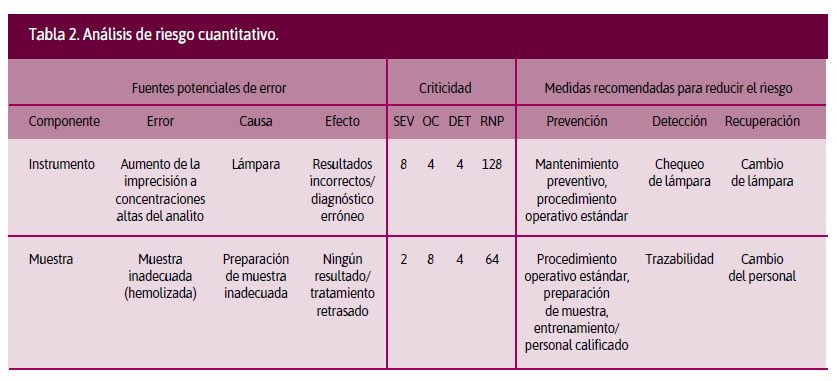
A este análisis cuantitativo se le conoce como Failure modes and effects analysis (FMEA o Análisis de los modos de falla y sus efectos potenciales). El modo de falla se define como la manera en que se observa un fracaso, describiendo cómo se produce y cuál podría ser su impacto en la operación del instrumento o proceso.6
GESTIÓN DE RIESGOS EN SALUD
En el año 2011, en el foro nacional de calidad de los EE. UU., se incluyeron dos nuevos eventos graves, relacionados con los laboratorios clínicos:
1. Que se produzca la muerte o una lesión grave del paciente, como resultado de la pérdida irrecuperable de un espécimen biológico irreemplazable.
2. Que se produzca la muerte o una lesión grave del paciente, como resultado de la falta de comunicación de un resultado crítico de laboratorio (también incluyen resultado de radiología y anatomía patológica).7
Hoy en día, esta visión del manejo del riesgo en los servicios de salud, sobretodo en establecimientos de salud públicos, es un tema complejo y de prioritaria intervención. A veces se priorizan ciertos indicadores de gestión, buscando incrementar la cobertura de los tamizajes; pero muchas veces, en el afán por cumplir las metas mensuales, omitimos completar esta búsqueda con la vigilancia final de los pacientes tamizados y diagnosticados con tal o cual enfermedad. Del mismo modo, no se fortalecen los controles óptimos para la comunicación final de los resultados a los pacientes. Por ejemplo, en muchos hospitales públicos contamos con sistemas de información hospitalaria (HIS), donde los diferentes servicios o UPSS (unidades prestadoras de servicios de salud), como los laboratorios clínicos, adaptan su diseño según sus limitaciones y necesidades. Lo ideal es que tengamos un sistema de información para laboratorio (LIS); esta es una herramienta que permitiría, por ejemplo, obtener alarmas diarias y radares, para realizar el seguimiento de resultados críticos. Los gestores en los establecimientos de salud deberían, por lo menos, hacer un análisis de riesgo cuantitativo, una vez al año, priorizando los peligros y riesgos según su diagnóstico situacional. De esta manera se podrá construir el árbol de fallas, diagramas de flujo y la matriz FMEA.8
En EE. UU., la Administración de Salud de los Veteranos (Veterans Health Administration) aplica este análisis de GR en sus centros de salud, bajo una metodología denominada HFMEA (Healthcare Failure Mode and Effect Analysis), que es una modificación del FMEA. Describiéndola como una herramienta invaluable dentro de la seguridad del paciente. Desde su implementación, se han completado cientos de HFMEA, lo que ha permitido realizar mejoras en el sistema sanitario de los veteranos.3 La Joint Commission International (JCI) aplica la metodología denominada FMEA in health care: proactive risk reduction-2010, para su aplicación en el entorno sanitario.9
Según cifras del Medicare, uno de cada siete pacientes experimenta un error médico. Los errores médicos pueden ocurrir en cualquier parte del sistema de atención médica: en hospitales, clínicas, centros de cirugía, consultorios médicos, farmacias y en el hogar de los pacientes. Estos errores pueden estar relacionados con medicamentos, cirugías, diagnósticos, equipos o resultados de laboratorio.10
Considerando que en el sistema de salud de los EE. UU. las regulaciones son más exigentes y, a pesar de ello, existen errores médicos; en nuestros establecimientos de salud públicos y privados, ¿cuántos errores médicos existirán?
GESTIÓN DE RIESGOS EN LOS LABORATORIOS CLÍNICOS
Antecedentes
En el año 1988, CLIA (Clinical Laboratory Improvement Amendments) estableció que para el control de calidad se utilizarían dos niveles de control (normal y patológico) para las pruebas cuantitativas y dos niveles para las pruebas cualitativas (positivo y negativo). En 1991 se elaboró la directriz "Control estadístico de calidad", para mediciones cuantitativas: procedimientos, principios y definiciones (C24-A), aprobada por el CLSI, que evalúa parámetros de precisión y exactitud. En 2004 se realizó una modificación conocida como "Control de calidad equivalente", pero tuvo muchas críticas. Por ello, el CLSI en 2005 conformó un comité para que elabore una directriz para la planificación del control de calidad basado en la gestión de riesgos. En 2011, el CLSI publicó el documento EP23-A, sobre el control de calidad basado en el laboratorio basado en la gestión de riesgos. Este protocolo fue reconocido por los Centers for Medicare & Medicaid Services (CMS). En 2013, CMS establecen que a partir del 1 de enero de 2016 la norma EP23-A debe ser implementada en los laboratorios clínicos de los EE. UU. y desarrollarse un plan individualizado de control de calidad (IQCP).11,12
Normas internacionales relacionadas con la GR
Las tres principales normas relacionadas con el manejo de la GR en los laboratorios clínicos provienen de la ISO 14971: Medical devicesApplication of risk management to medical devices, la EP18A2: Risk management techniques to identify and control laboratory error sources (del CLSI), generalmente dirigida a los fabricantes, y la EP23-A: Laboratory quality control based on risk management (del CLSI), aplicada específicamente en los laboratorios clínicos.
-
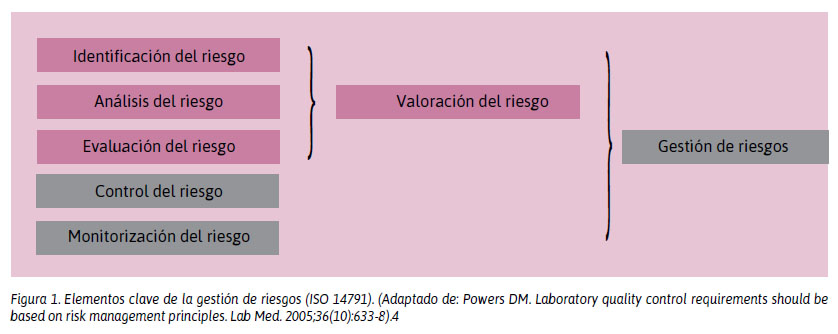
Norma ISO 14971: Medical devices Application of risk management to medical devices (Aplicación de la gestión de riesgos a los productos sanitarios). Describe a la GR como la aplicación sistemática de políticas, procedimientos y prácticas a las tareas de análisis, evaluación, control y seguimiento del riesgo. Hay que anticiparse a lo que podría salir mal, evaluar la frecuencia de ocurrencia de estos errores, así como las consecuencias o gravedad del daño y, finalmente, lo que se puede hacer para reducir el riesgo de daño. Es un concepto nuevo para los laboratorios clínicos. En la Figura 1 se detallan los elementos clave de la gestión de riesgos, según la norma ISO 14971.13
-
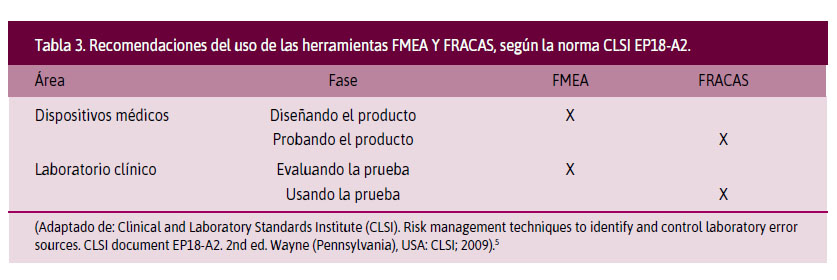
Norma CLSI EP18-A2: Risk management techniques to identify and control laboratory error sources (Técnicas de gestión de riesgos para identificar y controlar las fuentes de error en el laboratorio). Describe las herramientas de la GR. Estas deberán evaluarse en conjunto para su aplicación. Propone el uso de las herramientas Failure modes and effect analysis (FMEA o Análisis de los modos de falla y sus efectos potenciales) y Fault tree analysis (FTA o Análisis del árbol de falla) al inicio de la evaluación de un nuevo ensayo o instrumento. Sugiere consultar al fabricante sobre los riesgos conocidos y evaluar los riesgos específicos del laboratorio. Posteriormente, propone la aplicación del Failure reporting and corrective action system (FRACAS o Sistema de informe de fallas y acciones correctivas), donde se detallan las fallas producidas y las medidas de control empleadas para corregirlas. En la Tabla 3, se observan las recomendaciones de aplicación de las herramientas para la evaluación o uso de las pruebas en un laboratorio clínico, según la norma CLSI EP18-A2.5
-
Norma CLSI EP23-A: Laboratory Quality Control Based on Risk Management (Control de calidad en el laboratorio basado en la gestión de riesgos). Describe cómo desarrollar y mantener un plan de control de calidad para las pruebas de laboratorio, basado en los principios de la gestión de riesgos industriales.
Este plan debe detectar las deficiencias en las etapas preanalítica, analítica y posanalítica, delineando acciones específicas para la detección, prevención y control de fallas que puedan ocasionar un daño al paciente. En un laboratorio siempre existe algún riesgo de error durante el proceso de una prueba. Por lo tanto, la valoración del riesgo deberá priorizarse en cinco componentes vitales: muestras, condiciones ambientales, reactivos, instrumento o analizador y operador.14 Los pasos para la elaboración del plan control de calidad en el laboratorio se resumen en la Figura 2.
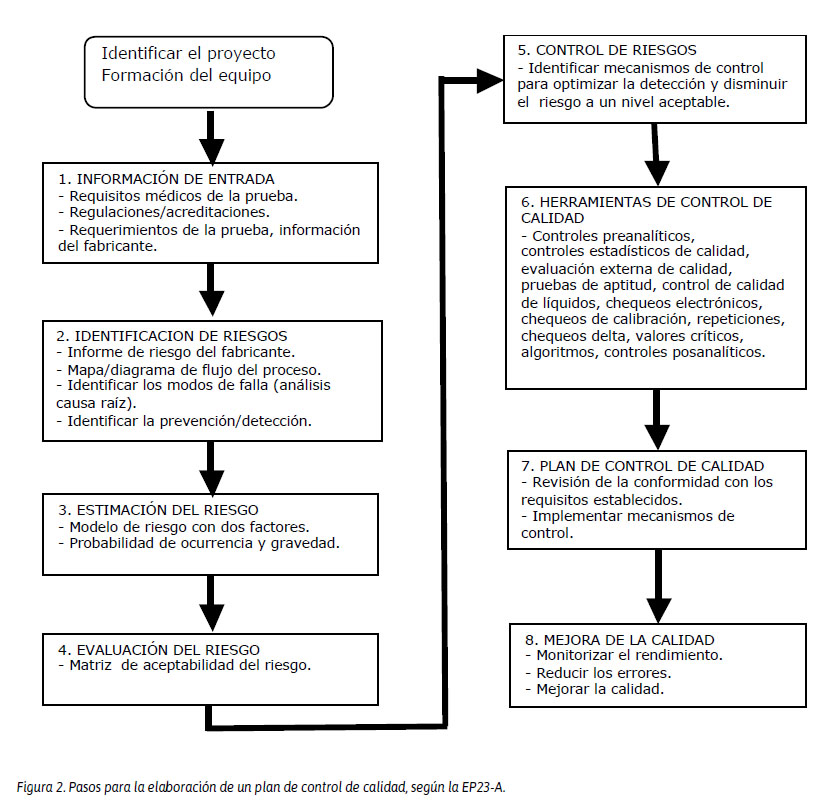
EVALUACIÓN DEL PROCESO ANALÍTICO Y DEL RIESGO
En todo proceso analítico existen posibles fallas o errores que deben ser evaluados y medirse la criticidad de los mismos, seleccionar las medidas de control y mantener niveles de riesgo clínicamente aceptables. Por ejemplo, en procesos analíticos donde se tiene control de calidad de líquidos, que es lo más frecuente en las plataformas analíticas hoy en día, un control analítico interno correctamente monitorizado podrá supervisar el rendimiento de un sistema de medición, permitiendo detectar los peligros que podrían limitar la utilidad de la prueba para su uso clínico previsto, se evaluará el rendimiento de los métodos (tipo de regla de control, número de controles, número de corridas analíticas, otros) y, finalmente, se podrá medir la probabilidad de detección de error y de falsos rechazos. Sin embargo, estos controles de calidad de líquidos detectarán errores causados por mala técnica: pipeteo, diluciones o preparación inadecuada del reactivo; pero no detectarán defectos preanalíticos o posanalíticos o errores aleatorios impredecibles, como: presencia de coágulos, fibrina, burbujas, lipemia o hemólisis.
Afortunadamente, con el avance de la tecnología, observamos con más frecuencia equipos automatizados para el proceso analítico y paquetes de reactivos con las fechas de vencimiento impresos en códigos de barra, lo que evita que el equipo lo utilice después de su caducidad; analizadores que detectan hemólisis, lipemia, ictericia o liberan alarmas para la interpretación técnica de la prueba. A esto se adicionan sistemas informáticos de laboratorio con alarmas o radares para detectar valores críticos, definir promedios históricos de cada paciente y evaluar diferencias significativas. La evaluación externa de calidad o prueba de aptitud es otro proceso de control que el laboratorio puede usar para garantizar el rendimiento del sistema de prueba. Estas fortalezas permiten a los laboratorios poder liberar resultados confiables, de modo que el médico en la consulta clínica pueda definir diagnósticos adecuados para beneficio de los pacientes.11
SEGUIMIENTO DEL PLAN DE CONTROL DE CALIDAD INDIVIDUALIZADO
Bajo este análisis, ante la identificación de los peligros y riesgos analíticos, se deben establecer las medidas de control para mitigar cada uno de ellos. Este plan implementa y monitoriza la efectividad (errores detectados y evitados), analiza las quejas y se previene la recurrencia del error en el futuro. Posteriormente, se revalúan los nuevos errores, los de mayor frecuencia e impacto en la salud de los pacientes y se realizan las correcciones necesarias para mantener un riesgo clínicamente aceptable.
En una reciente publicación, James Westgard efectúa algunas modificaciones sustanciales al plan de control de calidad individualizado propuesto por la norma CLSI EP23-A: establece un análisis del riesgo, al inicio cualitativo y posteriormente cuantitativo, usando herramientas como el FRACAS y las herramientas de calidad propuestas por él, como el "Six sigma", con lo que fortalece este plan.3 Inclusive, demuestra como ejemplo la experiencia de la aplicación de este análisis de riesgo, calculando el daño, la probabilidad de ocurrencia y la capacidad de detección (FMEA), con el sistema "Six sigma" en un laboratorio de Portugal (EndocLab).15
ÁREAS DE INCERTIDUMBRE
La frecuencia de los errores observados a lo largo del proceso analítico son: preanalíticos, 60%; analíticos, 15% y posanalíticos, 25%.16 Pero estos resultados hoy en día están cambiando, debido a que los profesionales de laboratorio clínico están saliendo de su cuatro paredes y entienden que existen dos componentes adicionales que permiten cerrar el círculo cerebro-cerebro. Esto significa que los errores pueden partir desde la etapa prepreanalítica, cuando el médico solicita al paciente, después de una evaluación médica, los análisis necesarios para diagnosticar o monitorizar su enfermedad. Posterior a ello, el paciente acudirá a su toma de muestra (etapa preanalítica), se obtiene el espécimen de estudio y este pasa a ser analizado (etapa analítica); después, se valida y libera el resultado para su ingreso al sistema informático de laboratorio (etapa posanalítica). Con este resultado, el paciente acudirá a su médico para la evaluación respectiva (etapa posposanalítica), culminando con esto todo el ciclo cerebro-cerebro, donde el médico que solicitó el análisis evaluará el resultado y brindará un diagnóstico y/o tratamiento.17 En la Figura 3 se detallan las etapas del proceso analítico de la prueba.
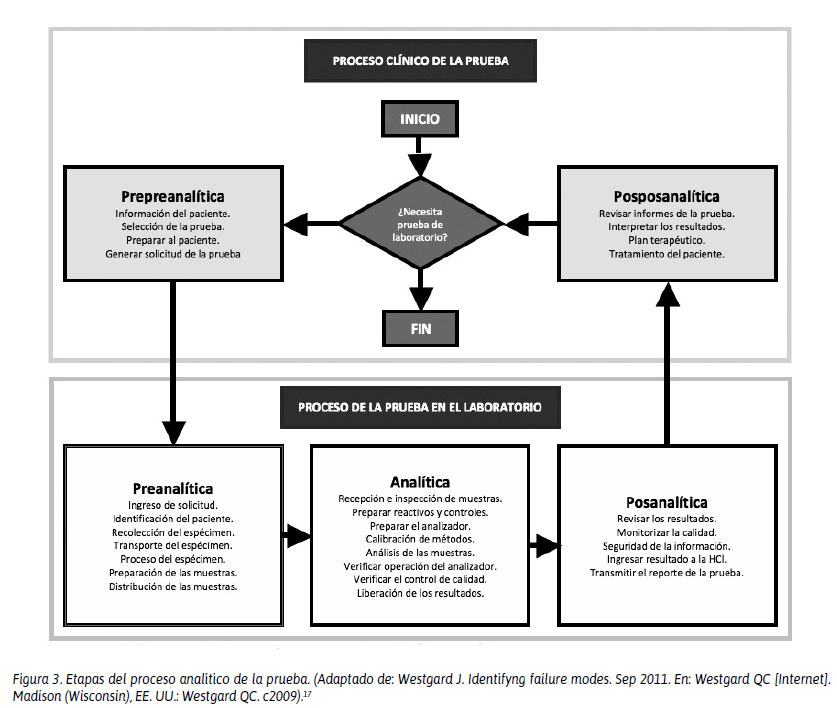
Con relación al impulso del plan de control de calidad individualizado (Individualized Quality Control Plan -IQCP), propuesto por CMS y que entra en vigencia desde enero de 2016, Westgard refiere que su implementación se está acelerando actualmente en los EE. UU. Las empresas están desarrollando iniciativas de educación, nuevos programas informáticos, seminarios, talleres y tutoriales en línea. Pero, ¿alguien realmente entiende lo que significa este plan?, ¿alguien sabe lo que significa la "P"?, ¿problema?, ¿plan?, ¿placebo? El autor refiere que este plan tiene matices por considerar, porque otorga al laboratorio clínico la facultad de implementarlo sobre la base de sus necesidades, sin priorizar las necesidades del paciente. Actualmente, los fabricantes no quieren invertir más en dispositivos de calidad y los sistemas de salud buscan el ahorro de costos. Por lo tanto, las evaluaciones cualitativas del riesgo pueden poner en peligro el control de calidad, priorizando a las herramientas de gestión de riesgos sobre la calidad requerida para los pacientes.18
Otro estudio refiere que la aplicación de listas de verificación es un tipo débil de barrera de seguridad, pues son vulnerables a la normalización de la desviación, especialmente en aquellos puntos que no se perciben como importantes para todos los usuarios. Además, estas listas de verificación proporcionan ganancias en seguridad; pero esos logros amenazan la eficiencia, lo que resulta en la transformación de ganancias de seguridad por ganancias de producción. Estas barreras contra el daño al paciente podrían ser percibidos como un reemplazo de la lista de verificación y, por lo tanto, ser ignorados con el fin de mejorar la producción.19
Sobre el uso de la herramienta FMEA, varios estudios refieren que su uso en la asistencia sanitaria se asocia con la falta de estandarización, en especial sobre las escalas de puntuación que utilizan y cómo se priorizarán los riesgos. Su aplicación es subjetiva y el uso de puntuaciones numéricas da una impresión injustificada de objetividad y precisión. Es sorprendente que esta herramienta, de uso tan común y ampliamente empleada dentro del cuidado de la salud, no parece que tenga pruebas de que sus resultados sean válidos y fiables, además de requerir mucho tiempo del personal para su elaboración.20-24
INSTRUMENTO DE SUPERVISIÓN DE LAS UPSS DE PATOLOGÍA CLÍNICA POR SUSALUD
En enero de 2015, la Superintendencia Nacional de Salud (Susalud), según la Resolución de Superintendencia N.° 006-2015-Susalud/S, aprobó el nuevo instrumento de supervisión selectiva, aplicable a las unidades productoras de servicios de salud (UPSS) de patología clínica.25 Este nuevo instrumento está siendo aplicado en las UPSS de patología clínica públicas y privadas en nuestro país. Después de que los laboratorios son supervisados, Susalud remite un porcentaje final de riesgo, con relación al cumplimiento del número de verificadores: menor de 60% de cumplimiento, alto riesgo; entre 60 y 80%, moderado riesgo y mayor de 80%, bajo riesgo. Además, remite una matriz donde cada laboratorio debe elaborar un plan de mitigación de riesgos para la implementación de las mejoras a favor del levantamiento de las observaciones. A pesar de que incluyen términos como «mitigación del riesgo», la metodología no se relaciona con las herramientas establecidas para la gestión de riesgos, según las directrices de los laboratorios, actualmente. Asimismo, los 65 verificadores que incluye el instrumento, en su mayoría, evalúan parámetros preanalíticos, pero no se toman en cuenta a los parámetros prepreanalíticos y posposanalíticos.
CONCLUSIONES
Las aerolíneas están actualizando sus mecanismos para la gestión de riesgos y evitar accidentes semejantes al sufrido por aerolínea Germanwings. En nuestro caso, los establecimientos de salud y sus laboratorios clínicos deberían empezar a aplicar las normas de control de calidad, como la EP23-A, basado en la gestión de riesgos, diseñar los análisis de gestión de riesgos, evaluar los peligros y sus riesgos, priorizarlos e intervenir sobre ellos, para el beneficio de los pacientes.
REFERENCIAS BIBLIOGRÁFICAS
1. Rosnoblet JF. German airbus crashes in French Alps with 150 dead, black box found [Internet]. Reuters. 24 mar 2015; Sect. Business [citado 1 jul 2015]; [aprox. 3 pantallas]. Disponible en: http://www.reuters.com/article/2015/03/25/us-france-crashairbus-lufthansa-idUSKBN0MK0ZP20150325.
2. International Organization for Standardization/International Electrotechnical Commission (ISO/IEC). Safety aspects-Guidelines for their inclusion in standards. ISO/IEC Guide 51:2014 [Internet]. Ginebra, Suiza: ISO/IEC; 2014 [citado 25 abr 2015]. Disponible en: http://isotc.iso. org/livelink/livelink/fetch/2000/2122/4230450/8389141/ ISO_IEC_Guide_51_2014%28E%29_-_Safety_ aspects_--_Guidelines_for_their_inclusion_in_standards. pdf?nodeid=8389248&vernum=-2
3. Westgard JO. Perspectives on quality control, risk management, and analytical quality management. Clin Lab Med. 2013;33:1-14.
4. Powers DM. Laboratory quality control requirements should be based on risk management principles. Lab Med. 2005;36:633-8.
5. Clinical and Laboratory Standards Institute (CLSI). Risk management techniques to identify and control laboratory error sources. CLSI document EP18-A2. 2nd ed.Wayne (Pennsylvania), USA: CLSI; 2009.
6. McDermott RE, Mikulak RJ, Beauregard MR.The basics of FMEA. 2nd ed. New York, USA: CRC Press; 2009.
7. Serious reportable events in healthcare-2011 update: a consensus report [Internet]. Washington, D.C., USA: National Quality Forum;2011 [citado 25 abr 2015].Disponible en:https:// www.qualityforum.org/Topics/SREs/List_of_SREs.aspx
8. Krouwer JS. Managing risk in hospitals using integrated fault trees and failure mode effects and criticality analysis.Washington, D. C.: USA:AACC Press; 2004.
9. Joint Commission International. Failure mode and effects analysis in health care: proactive risk reduction. 3th ed. Oak Brook (Illinois), USA: Joint Commission Resources; 2010.
10. U. S. Department of Health & Human Services, Agency for Healthcare Research and Quality. Veinte consejos para ayudar a evitar errores médicos [Internet]. Rockville (Maryland), EE. UU.: AHRQ; 2014[citado 25 may2015]. Disponible en: http://www.ahrq.gov/patients-consumers/care-planning/ errors/20tips/20tipssp.html
11. Njoroge SW, Nichols JH. Risk management in the clinical laboratory.Ann Lab Med. 2014;34(4):274-8.
12. U. S. Department of Health & Human Services, Centers for Medicare & Medicaid Services (CMS), Clinical Laboratory Improvement Amendments (CLIA). Individualized Quality Control Plan (IQCP) [Internet]. Baltimore (Maryland), USA: CMS [citado 15 may 2015]. Disponible en: https://www.cms. gov/regulations-and-guidance/legislation/CLIA/Individualized_ Quality_Control_Plan_IQCP.html
13. International Organization for Standardization (ISO). Medical devices - Application or risk management to medical devices. ISO 14971. Ginebra, Suiza: ISO; 2007.
14. Clinical and Laboratory Standards Institute (CLSI). Laboratory quality control based on risk management. CLSI document EP23-A. Wayne (Pennsylvania), USA: CLSI; 20 Wayne (Pennsylvania), USA: CLSI; 2011.
15. Westgard S. Prioritizing risk analysis quality control plans based on Sigma-metrics. Clin Lab Med. 2013;33(1):41-53.
16. Westgard J,Mercapide L,Sáez A,Porras A,Martínez O,Amaya E, et al. Cómo garantizar la calidad analítica. Rev Mex Patol Clin. 2010;57(4):179-89.
17. Westgard J. Identifyng failure modes. Sep 2011 [citado 10 jun 2015]. En: Westgard QC [Internet]. Madison (Wisconsin), EE. UU.:Westgard QC.c2009 - .[aprox.5 pantallas].Disponible en: https://www.westgard.com/identifying-failure-modes.htm.
18. Westgard J.IQCP:Does the P stand for Placebo? Jun 2014 [citado 15 jun 2015]. En:Westgard QC [Internet]. Madison (Wisconsin), EE.UU.:Westgard QC.c2009 - .[aprox.6 pantallas].Disponible en: https://www.westgard.com/iqcp-placebo.htm
19. Rydenfält C, Ek Å, Larsson PA. Safety checklist compliance and a false sense of safety: new directions for research. BMJ Qual Saf. 2014;23(3):183-6.
20. Bowles JB; South Carolina University. An assessment of RPN prioritization in a failure modes effects and criticality analysis. In: Annual Reliability and Maintainability Symposium, 2003 Annual. Columbia, South Carolina, EE. UU.: IEEE; 2003. p.380-6.
21. Shebl NA,Franklin BD,Barber N.Failure mode and effects analysis outputs: are they valid? BMC Health Serv Res. 2012;12:150.
22. Dean Franklin B, Shebl NA, Barber N. Failure mode and effects analysis: too little for too much? BMJ Qual Saf. 2012;21:607-11.
23. Shebl NA, Franklin BD, Barber N. Is failure mode and effect analysis reliable? J Patient Saf. 2009;5(2):86-94.
24. Westgard J. Risk Analysis: Is it Reliable? Is it Valid? Is it too little for too much? Sep 2012 [citado 15 jun 2015]. En:Westgard QC [Internet].Madison (Wisconsin),EE.UU.:Westgard QC.c2009 - . [aprox. 8 pantallas]. Disponible en: https://www.westgard.com/fmea-reliability.htm.
25. República del Perú,Superintendencia Nacional de Salud (Susalud). Resolución de Superintendencia N.° 006-2015-Susalud/S, Instrumento de Supervisión Selectiva de IPRESS aplicable a las Unidades Productoras de Servicios de Salud Patología Clínica de las Ipress públicas y privadas (promulgado 9 de enero de 2015). Lima, Perú: El Peruano, Normas Legales. 16 ene 2015. p. 544853
Correspondencia
Dr. Luis Figueroa Montes
patologoclinico@gmail.com
Conflictos de interés
El autor declara no tener conflictos de interés durante el planteamiento, ejecución de la investigación y la elaboración del artículo para su publicación.
Fecha de recepción: 20 de julio de 2015
Fecha de aceptación: 23 de noviembre de 2015
