










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Médica Peruana
On-line version ISSN 1728-5917
Acta méd. Peru vol.34 no.2 Lima Apr. 2017
REPORTE DE CASO
Sarcoma histiocítico: presentación de un caso probable
Histiocytic sarcoma: Presentation of a probable case
Luis F. Duran-Gutierrez1,4, Clara Alarcon-Gil2, Ernesto F. Benavides-Lopez1,2, Luis C. Alvarez-Perdomo1,4, Jaime M. Jimenez-Duarte3, Stephany Correa- Florez4, Alvaro Mondragon-Cardona1,4, Cristhian F. Ramirez-Ramos1,4
1 Servicio Medicina Interna, Universidad Surcolombiana, Hospital Hernando Moncaleano Perdomo. Neiva, Colombia.
2 Servicio de Hematología y Oncología, Hospital Hernando Moncaleano Perdomo. Neiva, Colombia.
3 Servicio de Patología, Hospital Hernando Moncaleano Perdomo. Neiva, Colombia.
4 Grupo de investigación MI-Dneuropsy. Semillero de investigación de medicina interna (SIMI-USCO) Universidad Surcolombiana. Neiva, Colombia.
RESUMEN
El sarcoma histiocítico es un desorden de células no Langerhans extremadamente raro con un curso clínico agresivo y limitadas opciones de tratamiento. Se presenta el caso de un paciente adulto joven de género masculino, con cuadro febril prolongado, compromiso hematológico persistente, hepatoesplenomegalia y linfadenopatias generalizadas con curso clínico fatal, en quien por las características clínicas, paraclínicas y de estudios histopatológicos como de inmunohistoquímica se llegó a dicha presunción diagnostica.
Palabras clave: Sarcoma histiocítico; Pancitopenia; Linfoma; Linfadenopatía; Colombia (fuente: DeCS BIREME).
ABSTRACT
Histiocytic sarcoma is an extremely rare non-Langerhans cell disorder with an aggressive clinical course and limited options for therapy. We present the case of a young adult male patient, with prolonged fever, persistent hematological involvement, hepatosplenomegaly, and generalized lymphadenopathy with a fatal clinical course, in whom we arrived to this presumptive diagnosis considering the clinical, paraclinical, and histopathological characteristics, as well as immunohistochemistry.
Keywords: Histiocytic sarcoma; Pancytopenia; Lymphoma; Lymphadenopathy; Colombia (source: MeSH NLM).
INTRODUCCIÓN
El sarcoma histiocítico es una malignidad rara, correspondiendo a aproximadamente 1% de todas las neoplasias de origen hematopoyético (1). Afecta a todos los grupos etarios, con un pico entre los 45 y 55 años de edad (2). Su etiología aun es desconocida y tiene características distintas desde el punto de vista biológico, con cuadros clínicos variados y sin tratamiento estandarizado. Datos genómicos y de inmunofenotipificación limitados, aunados a la superposición con características histológicas con otras formas de neoplasias hematológicas en especial linfomas no Hodgkin han generado muchas controversias en cuanto a su diagnóstico. Sin embargo, en los últimos años y con los resultados de estudios de inmunohistoquímica se pudo realizar la identificación consistente como una entidad única distinta.
En el año 2001, la clasificación del sarcoma histiocítico dado por la Organización Mundial de la Salud (OMS) se basó en la ausencia de reordenamientos clonales del receptor de células T o B. En el 2008 la OMS reclasificó al sarcoma histiocítico para incluir los rearreglos del receptor de células T y de las cadenas pesadas de las inmunoglobulinas; entonces estos casos se agruparon como formas transdiferenciadas de otras líneas hematopoyéticas (2). En la literatura médica se encuentra una clara asociación entre la mencionada entidad y varias malignidades, en especial aquellas que tienen un origen en células B; esta ocurre como un evento secundario luego de las enfermedades hematolinfoides y se ha demostrado que alberga las mismas anormalidades moleculares o citogenéticas de la malignidad primaria. Los reportes incluyen asociación con linfoma folicular (3), leucemia monocítica aguda (4), leucemia de células peludas (5), linfoma de células del manto (6), linfoma tipo MALT (7), linfoma difuso de células grande B (8), linfoma linfoblástico agudo (9), leucemia linfocítica crónica (10), leucemia mielomonocítica crónica (11), tumor de células germinales (12), mielofibrosis idiopática (13) y leucemia mieloide crónica.
Se presenta el caso de un paciente adulto joven en cuyo abordaje diagnóstico y estudios de inmunohistoquímica en médula ósea se considera posible sarcoma histiocítico.
REPORTE DE CASO
Hombre de 21 años, agricultor, procedente de zona rural del sur de Colombia ubicada a 941 m de altitud. Ingresó a establecimiento sanitario por cuadro clínico de quince días consistente en picos febriles no cuantificados de predominio nocturno asociados a cefalea, mialgias, artralgias, tos seca y deposiciones diarreicas líquidas sin moco ni sangre. Como antecedentes de importancia bebía agua no tratada y vivía con animales de granja. Estudios realizados en primer nivel de atención documentaron leucopenia, trombocitopenia y reactantes de fase aguda (proteína C reactiva, PCR) positivos (Tabla 1), radiografía de tórax y ecografía abdominal en límites normales. En tal situación, se le administró cefalosporina de tercera generación.
A pesar de manejo instaurado, el paciente persistió febril, con leve tinte ictérico, adenopatías en cuello, región supraclavicular e inguinal y abdomen con hepatoesplenomegalia; ante lo cual se consideró síndrome ictérico-hemorrágico con alta probabilidad de leptospirosis por lo que se lo remitió a institución de mayor nivel. Se realizó tomografía abdominal simple y contrastada, tomografía de tórax, ecocardiograma transtorácico, tamizaje para virus hepatotropos así como causas endémicas de fiebre persistente; los resultados de todas estas pruebas se encontraban dentro de límites normales.
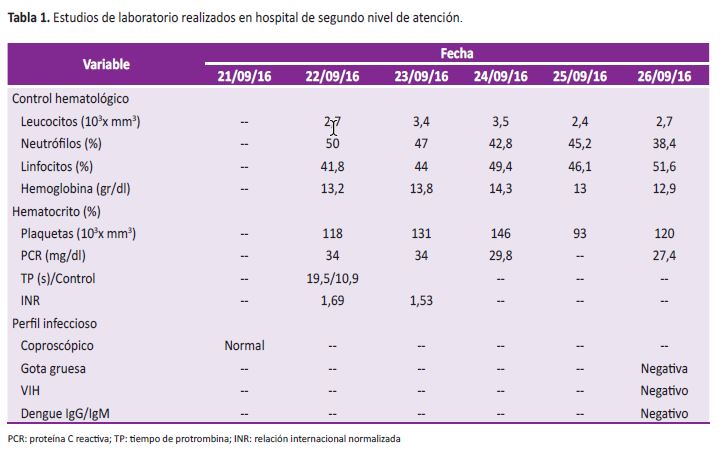
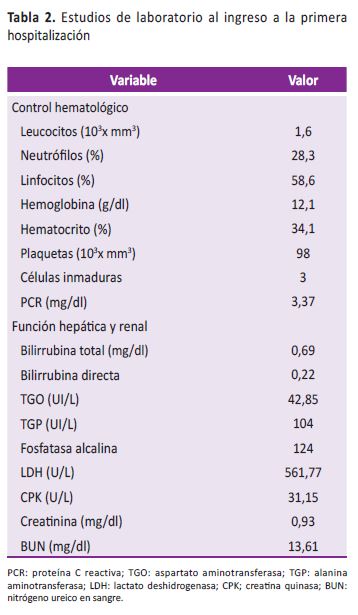
Durante observación persistió febril, con mejoría lenta de cuadro diarreico, evolución paraclínica con función renal conservada, transaminasas elevadas, controles hematológicos con progresión a pancitopenia y presencia de células inmaduras (Tabla 2) a pesar de que se amplió espectro de cubrimiento antibiótico con tetraciclina.
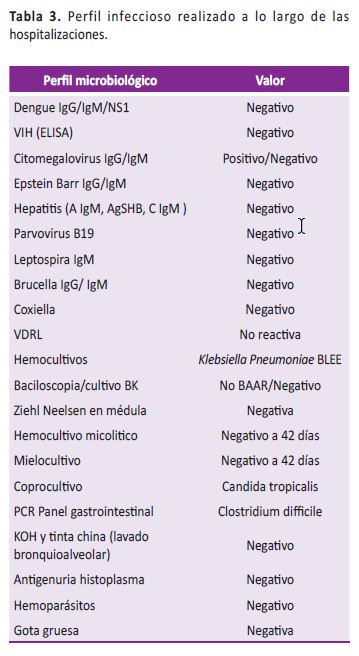
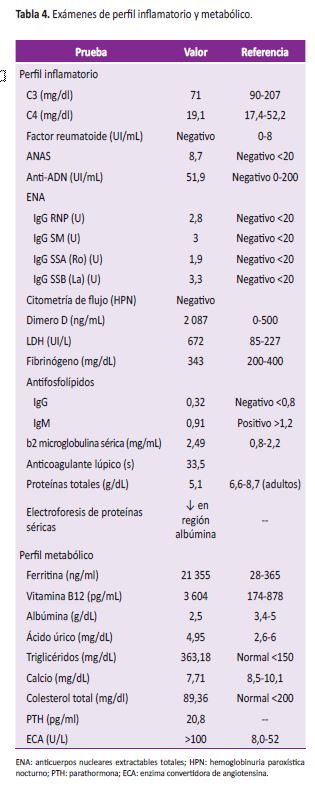
Se consideró pertinente descartar proceso neoplásico hematológico. Se realizó aspirado y biopsia de médula ósea con estudio de citometría de flujo. El resultado de la biopsia de médula ósea mostraba inflamación granulomatosa, por lo que se consideró tuberculosis dentro de posibles etiologías y se inició manejo antifímico. Ante falta de claridad diagnóstica, se complementaron estudios para causas infecciosas (Tabla 3), ajustadas a entorno epidemiológico, así como metabólicas e inflamatorias, como de autoinmunidad sin resultados concluyentes (Tabla 4). Adicionalmente, y por el tipo de inflamación a nivel de médula ósea, se tomaron biopsias de hígado y bazo cuyos reportes mostraron histiocitosis reactiva y hematopoyesis extramedular. Se estudiaron además muestras de epiplón, líquido peritoneal y peritoneo parietal cuyos estudios finalmente no tendrían lesiones destacables. El estudio de fibrobroncoscopia más lavado broncoalveolar no evidenció bacilos acido-alcohol resistente ni otras anormalidades de interés.
Tras dos meses de observación hospitalaria, el paciente persistía con fiebre, con difícil control de episodios diarreicos y dolor abdominal persistente. La evolución clínica fue hacia el franco deterioro, presentó bacteriemia por Klebsiella pneumoniae productora de betalactamasa de amplio espectro; una vez se logró estabilizar el paciente se realizó nuevo aspirado de médula ósea para estudio de inmunohistoquímica, además se amplió perfil infeccioso con para infección por virus Epstein- Barr; citomegalovirus y parvovirus. Se iniciaron pulsos de metilprednisolona y se suspendió manejo antituberculoso que se administró durante aproximadamente dos meses. Los resultados de la segunda biopsia de médula ósea junto a los estudios de inmunohistoquímica permitieron aclarar el diagnóstico, concluyendo que por el alto índice de proliferación de población histiocitaria el diagnóstico era compatible con afectación de médula por sarcoma histiocítico (Figura 1). Finalmente, el paciente progresó al deterioro neurológico con parálisis completa del III par craneal izquierdo sin demostrarse alteraciones en tomografía cerebral, presentó neutropenia febril absoluta y nueva bacteriemia por Klebsiella pneumoniae sin respuesta a manejo antibiótico, compromiso de su estado de conciencia con rápida progresión a estupor y falla ventilatoria sin respuesta al manejo y muerte en las 24 horas siguientes.
DISCUSIÓN
Presentamos el caso de un paciente masculino en la tercera década de la vida quien presenta síndrome febril de origen desconocido y pancitopenia, múltiples complicaciones infecciosas, deterioro neurológico y muerte.
El sarcoma histiocítico fue descrito por primea vez en 1970 por Mathe y colaboradores (14) como tumores compuestos de células libres las cuales tenían una morfología variable con abundante citoplasma eosinofílico en las cuales el núcleo estaba a menudo distorsionado y monocitoide. Esta entidad corresponde a un tipo de desorden histiocitario que pertenece a un grupo de enfermedades con clínica y pronósticos heterogéneos. Las neoplasias histiocitarias se dividen en dos grupos: histiocitosis de células de Langerhans e histiocitosis no Langerhans (15), perteneciendo el sarcoma histiocítico a éste último. Se define por la acumulación de histiocitos, células derivadas de monocitos de la médula ósea que migran desde la sangre periférica a diferentes tejidos, aunque la proliferación local también se ha descrito (16). Representa menos del 1% de todos los procesos malignos del sistema hematopoyético (1). Es extremadamente rara y con un curso agresivo, los estudios de casos con criterios actuales para diagnósticos más amplios incluyen de seis a siete casos. Antes era desconocido si representaba un cuadro autoinmune o un tipo de neoplasia, actualmente se reconoce como una neoplasia mieloide (15). Su etiología es desconocida, se ha visto algunos casos asociados en pacientes con tumores mediastinales de células germinales ya que en presencia de estos tumores hay células pluripotenciales capaces de dar lugar a colonias de macrófagos y otros tipos de células (17). Se han establecido asociaciones también con linfomas foliculares, síndromes mielodisplásicos y leucemia linfoblástica aguda (18). De hecho, se ha descrito como un proceso maligno secundario en múltiples casos (19).
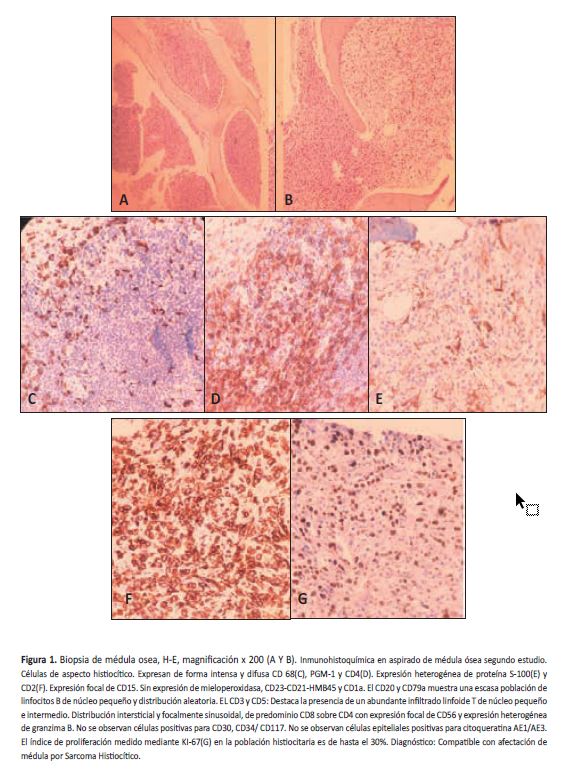
Se ha reportado en todos los grupos de edad, pero se ha visto con mayor frecuencia en adultos (promedio de 46-55 años) (18,20). Algunos estudios indican predilección por el sexo masculino (20,21), sin embargo, otros no asocian riesgo respecto al sexo. La forma de presentación usual son tumores extranodales multifocales o únicos principalmente en intestino, piel o tejidos blandos (22); hay también descripciones de linfadeponatías difusas y múltiples sitios de compromiso, en general estos pacientes con enfermedad multifocal tienen un peor pronóstico (23). En nuestro caso, el paciente presentaba compromiso de médula ósea, ganglios linfáticos y hepatoesplenomegalia, lo cual puede explicar lo corta de su sobrevida.
Los síntomas sistémicos como la fiebre y pérdida de peso son frecuentes y los síntomas de compresión de órganos vitales (intestino delgado) pueden ocurrir. El compromiso dérmico incluye rash e innumerables tumores en múltiples áreas corporales. Las citopenias son vistas en el 30% de los casos (24).
El diagnóstico por inmunohistoquímica se reconoce como parte fundamental de la identificación de esta patología, se incluyen como marcadores positivos en pacientes con sarcoma histiocítico el CD163, CD68 y lisozima, con negatividad para langerina y CD1a que podría sugerir un proceso neoplásico de tipo de células de Langerhans, así como para CD21, CD35, CD33. El marcador ki67 se describe como variable. También se ha descrito al ser un desorden histiocitario no Langerhans como inmunorreactivo para factor XIIIa y CD14, algunos expresan S100 (15). La citometría de flujo tiene múltiples reportes en la literatura donde reconocen su importancia e incluso se han descrito casos de su utilización como único orientador hacia este diagnóstico (18,25). Además de lo anterior es menester realizar estudio de PCR para reordenamiento de receptor de células T y descartar una neoplasia de este linaje, siendo uno de los requisitos para el diagnóstico según la clasificación mundial de la salud; desafortunadamente en nuestro paciente no pudimos contar con dicho reporte pese a solicitar el examen ante la dificultad de encontrar un centro que realizara el mismo. La observación microscópica puede evidenciar un citoplasma eosinofílico con vacuolas. Los núcleos son pleomórficos y pueden tener uno o más nucléolos. En ocasiones, las células pueden tener un aspecto xantomatoso (24).
No se dispone de un protocolo estándar para manejo de esta enfermedad debido a la rareza de la misma. Los pacientes usualmente se presentan con enfermedad diseminada y a menudo con pobre pronostico (24). Hay pocos reportes de tratamiento exitoso en adultos, en población pediátrica existen reportes de buena respuesta con quimioterapia (25,26).
El tratamiento depende de si la afectación es focal o multifocal. En los casos focales, la resección del tumor con radiación coadyuvante demuestra beneficios importantes (22). Cuando se ha utilizado esquema de quimioterapia, el esquema CHOP (ciclofosfamida, doxorrubicina, vincristina, prednisona) es el más difundido; sin embargo, solo 40-45% de los pacientes responden a este manejo (27). Por otro lado, este tipo de manejo presenta una mejor respuesta en casos focales, en casos multifocales como el de nuestro paciente el tratamiento ideal aún no está claro. De manera reciente el etopósido fue adicionado al régimen CHOP para tratar esta neoplasia como una forma agresiva de linfoma. Nuevas aproximaciones en el manejo han planteado uso de talidomida (28), alemtuzumab (29) y vemnurafenib (30,31) en casos de pocos pacientes con buenas respuestas terapéuticas. Así mismo otras aproximaciones sugieren la identificación de receptores de factores de crecimiento específicos que pueden llevar a mejores resultados en algunos casos (32). En nuestro paciente no fue posible iniciar manejo quimioterápico por la rápida evolución fatal que presentó.
Ante el reporte de la primera biopsia de médula ósea de inflamación granulomatosa y se pensó inicialmente en la posibilidad de tuberculosis diseminada, una revisión posterior del estudio se evidencio que no correspondía a inflamación granulomatosa caseificante característica de la infección por micobacterias aunado a que todos los estudios para dicha infección fueron negativos (baciloscopías en esputo, estudios de lavado broncoalveolar, cultivo de tuberculosis y PCR en sangre para tuberculosis); vale mencionar que en la literatura existe un caso reportado de coexistencia de tuberculosis y sarcoma histiocítico (33), con algunas características similares a nuestro caso.
En general la tasa global de supervivencia no es mayor de dos años en los pacientes.
Hay que tener en cuenta que éste siempre es un diagnóstico de exclusión por la rareza de la enfermedad y con una presentación que obliga a realizar un amplio trabajo diagnostico por las múltiples etiologías incluidas en el diferencial.
Fuente de financiamiento: autofinanciado.
Conflictos de interés: Los autores declara no tener conflictos de interés.
REFERENCIAS BIBLIOGRÁFICAS
1. Machado ES, de Miranda AC, Escopelli T, Caron R, Escopelli AC. Histiocytic sarcoma. Rev Bras Hematol Hemoter. 2011;33(2):155-7. [ Links ]
2. Takahashi E, Nakamura S. Histiocytic sarcoma: an updated literature review based on the 2008 WHO classification. J Clin Exp Hematop. 2013;53(1):1-8. [ Links ]
3. Brunner P, Rufle A, Dirnhofer S, Lohri A, Willi N, Cathomas G, et al. Follicular lymphoma transformation into histiocytic sarcoma: indications for a common neoplastic progenitor. Leukemia. 2014;28(9):1937-40. [ Links ]
4. Zhao J, Niu X, Wang Z, Lu H, Lin X, Lu Q. Histiocytic sarcoma combined with acute monocytic leukemia: a case report. Diagn Pathol. 2015;10:110. [ Links ]
5. Michonneau D, Kaltenbach S, Derrieux C, Trinquand A, Brouzes C, Gibault L, et al. BRAF(V600E) mutation in a histiocytic sarcoma arising from hairy cell leukemia. J Clin Oncol. 2014;32(35):e117-21. [ Links ]
6. Hure MC, Elco CP, Ward D, Hutchinson L, Meng X, Dorfman DM, et al. Histiocytic sarcoma arising from clonally related mantle cell lymphoma. J Clin Oncol. 2012;30(5):e49-53. [ Links ]
7. Alvaro T, Bosch R, Salvado MT, Piris MA. True histiocytic lymphoma of the stomach associated with low-grade B-cell mucosa-associated lymphoid tissue (MALT)-type lymphoma. Am J Surg Pathol. 1996;20(11):1406-11. [ Links ]
8. Wang E, Papalas J, Hutchinson CB, Kulbacki E, Huang Q, Sebastian S, et al. Sequential development of histiocytic sarcoma and diffuse large b-cell lymphoma in a patient with a remote history of follicular lymphoma with genotypic evidence of a clonal relationship: a divergent (bilineal) neoplastic transformation of an indolent B-cell lymphoma in a single individual. Am J Surg Pathol. 2011;35(3):457-63. [ Links ]
9. McClure R, Khoury J, Feldman A, Ketterling R. Clonal relationship between precursor B-cell acute lymphoblastic leukemia and histiocytic sarcoma: a case report and discussion in the context of similar cases. Leuk Res. 2010;34(2):e71-3. [ Links ]
10. Shao H, Xi L, Raffeld M, Feldman AL, Ketterling RP, Knudson R, et al. Clonally related histiocytic/dendritic cell sarcoma and chronic lymphocytic leukemia/small lymphocytic lymphoma: a study of seven cases. Mod Pathol. 2011;24(11):1421-32. [ Links ]
11. Mori M, Matsushita A, Takiuchi Y, Arima H, Nagano S, Shimoji S, et al. Histiocytic sarcoma and underlying chronic myelomonocytic leukemia: a proposal for the developmental classification of histiocytic sarcoma. Int J Hematol. 2010;92(1):168-73. [ Links ]
12. Song SY, Ko YH, Ahn G. Mediastinal germ cell tumor associated with histiocytic sarcoma of spleen: case report of an unusual association. Int J Surg Pathol. 2005;13(3):299-303. [ Links ]
13. Fukunaga M, Kato H. Histiocytic sarcoma associated with idiopathic myelofibrosis. Arch Pathol Lab Med. 2004;128(10):1167-70. [ Links ]
14. Mathe G, Gerard-Marchant R, Texier JL, Schlumberger JR, Berumen L, Paintrand M. The two varieties of lymphoid tissue 'reticulosarcomas', histiocytic and histioblastic types. Br J Cancer. 1970;24(4):687-95. [ Links ]
15. Durham BH, Diamond EL, Abdel-Wahab O. Histiocytic neoplasms in the era of personalized genomic medicine. Curr Opin Hematol. 2016;23(4):416-25. [ Links ]
16. Alonso-Dominguez JM, Calbacho M, Talavera M, Villalon C, Abalo L, Garcia-Gutierrez JV, et al. Cytogenetics findings in a histiocytic sarcoma case. Case Rep Hematol. 2012;2012:428279. [ Links ]
17. Cudennec CA, Johnson GR. Presence of multipotential hemopoietic cells in teratocarcinoma cultures. J Embryol Exp Morphol. 1981;61:51-9. [ Links ]
18. Dalia S, Shao H, Sagatys E, Cualing H, Sokol L. Dendritic cell and histiocytic neoplasms: biology, diagnosis, and treatment. Cancer Control. 2014;21(4):290-300. [ Links ]
19. Ansari J, Naqash AR, Munker R, El-Osta H, Master S, Cotelingam JD, et al. Histiocytic sarcoma as a secondary malignancy: pathobiology, diagnosis, and treatment. Eur J Haematol. 2016;97(1):9-16. [ Links ]
20. Dalia S, Jaglal M, Chervenick P, Cualing H, Sokol L. Clinicopathologic characteristics and outcomes of histiocytic and dendritic cell neoplasms: the moffitt cancer center experience over the last twenty five years. Cancers (Basel). 2014;6(4):2275-95. [ Links ]
21. Gounder M, Desai V, Kuk D, Agaram N, Arcila M, Durham B, et al. Impact of surgery, radiation and systemic therapy on the outcomes of patients with dendritic cell and histiocytic sarcomas. Eur J Cancer. 2015;51(16):2413-22. [ Links ]
22. Hornick JL, Jaffe ES, Fletcher CD. Extranodal histiocytic sarcoma: clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Extranodal histiocytic sarcoma: clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. 2004;28(9):1133-44. [ Links ]
23. Gianotti F, Caputo R. Histiocytic syndromes: a review. J Am Acad Dermatol. 1985;13(3):383-404. [ Links ]
24. Pileri SA, Grogan TM, Harris NL, Banks P, Campo E, Chan JK, et al. Tumours of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology. 2002;41(1):1-29. [ Links ]
25. Heath J, Burgett S, Gaca A, Jaffe R, Wechsler D. Successful treatment of pediatric histiocytic sarcoma using abbreviated high-risk leukemia chemotherapy. Pediatr Blood Cancer. 2014;61(10):1874-6 [ Links ]
26. Iwabuchi H, Kawashima H, Umezu H, Takachi T, Imamura M, Saitoh A, et al. Successful treatment of histiocytic sarcoma with cladribine and high-dose cytosine arabinoside in a child. Int J Hematol. 2017 Feb 28. doi: 10.1007/s12185-017-2202-8. (Epub ahead of print) [ Links ]
27. Wang H, Zhang J, Tao Q, Bian H, Shen Y, Li Y, et al. Flow cytometry used to identify histiocytic sarcoma: A case report. Cytometry B Clin Cytom. 2016;90(6):546-50. [ Links ]
28. Abidi MH, Tove I, Ibrahim RB, Maria D, Peres E. Thalidomide for the treatment of histiocytic sarcoma after hematopoietic stem cell transplant. Am J Hematol. 2007;82(10):932-3. [ Links ]
29. Shukla N, Kobos R, Renaud T, Teruya-Feldstein J, Price A, McAllister- Lucas L, et al. Successful treatment of refractory metastatic histiocytic sarcoma with alemtuzumab. Cancer. 2012;118(15):3719-24 [ Links ]
30. Idbaih A, Mokhtari K, Emile JF, Galanaud D, Belaid H, de Bernard S, et al. Dramatic response of a BRAF V600E-mutated primary CNS histiocytic sarcoma to vemurafenib. Neurology. 2014;83(16):1478- 80. [ Links ]
31. Vaughn J, Freitag C, Hemminger J, Jones J. BRAF V600E expression in histiocytic sarcoma associated with splenic marginal zone lymphoma: a case report. J Med Case Rep. 2017 Apr 5;11(1):92. [ Links ]
32. Schlick K, Aigelsreiter A, Pichler M, Reitter S, Neumeister P, Hoefler G, et al. Histiocytic sarcoma – targeted therapy: novel therapeutic options? a series of 4 cases. Onkologie. 2012;35(7-8):447-50. [ Links ]
33. Aggarwal R, Rao S. Coexistence of tuberculosis with histiocytic sarcoma: A rare association. J Cancer Res Ther. 2015;11(3):658. [ Links ]
Correspondencia
Alvaro Mondragón Cardona
alvaromondragoncardona@gmail.com
Recibido: 29/03/2017
Arbitrado por pares
Aprobado: 07/06/2017
Citar como: Duran-Gutierrez LF, Gil-Alarcon CE, Benavides-Lopez EF, Alvarez-Perdomo LC, Jimenez- Duarte JM, Correa-Florez S, et al. Sarcoma histiocítico: presentación de un caso probable. Acta Med Peru. 2017;34(2):136-42
