










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa Médica Peruana
versión On-line ISSN 1728-5917
Acta méd. Peru vol.35 no.1 Lima ene. 2018
ARTÍCULO DE REVISIÓN
Factores de riesgo en las enfermedades genéticas
Risk factors in genetic diseases
Hugo H. Abarca Barriga1,2a, Miguel Chávez Pastor1,3, Milana Trubnykova1, Jorge E. La Serna-Infantes4b, Julio A. Poterico5b
1 Servicio de Genética y Errores Innatos del Metabolismo, Instituto Nacional de Salud del Niño. Lima, Perú.
2 Universidad Ricardo Palma. Lima, Perú.
3 Universidad Peruana Cayetano Heredia. Lima, Perú.
4 Servicio de Citogenética y Citopatología, Hospital Nacional Guillermo Almenara Irigoyen. Lima, Perú.
5 Unidad de Genética y Biología Molecular, Instituto Nacional de Enfermedades Neoplásicas. Lima, Perú.
a Magíster en Genética, b Médico Residente en genética médica
RESUMEN
Existen más de 10 000 enfermedades genéticas descritas en el mundo y afectan alrededor del 7% de la población mundial, causando alta morbimortalidad y costos para los sistemas de salud pública. Representan un reto diagnóstico por la variabilidad clínica y la necesidad de pruebas diagnósticas moleculares. En el Perú, son escasas las investigaciones respecto a estas condiciones y, aunque se ha promulgado la Ley de Enfermedades Huérfanas o Raras (Ley Nº 29698), no se han implementado estrategias sanitarias nacionales para el diagnóstico, manejo y prevención. La presente publicación tiene como objetivo describir los factores de riesgo más frecuentes, los cuales están relacionados a enfermedades o síndromes de etiología genética.
Palabras clave: Genética; Genética médica; Factores de riesgo; Enfermedades raras (fuente: DeCS BIREME).
ABSTRACT
More than 10,000 genetic diseases have been described, which affect approximately 7% of the whole world population, leading to high morbidity and mortality rates, as well as to elevated healthcare costs. The diagnosis of these conditions is a tough challenge, because of their clinical variability and the low availability of molecular diagnostic tests. There is little research performed in Peru dealing with these diseases, and although a ‘Bill for Orphan or Rare Diseases’ (Law N° 29698) has been recently issued, no national healthcare strategies have been implemented for the diagnosis, management, and prevention of genetic diseases. This paper aims to describe the risk factors that are more frequently related to diseases or syndromes with a genetic origin.
Keywords: Genetics; Genetics, medical; Risk factors; Rare diseases (source: MeSH NLM).
INTRODUCCIÓN
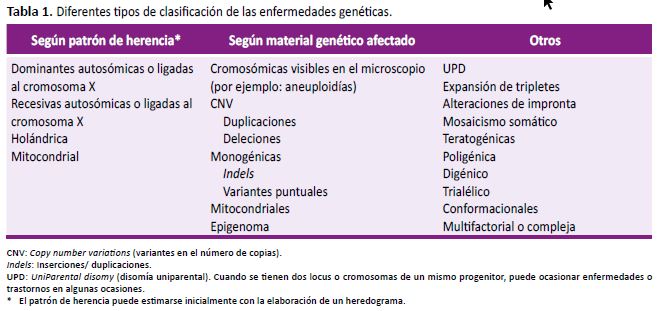
Las enfermedades o síndromes de etiología genética se encuentran, en su mayoría, clasificados y descritos en la base de datos OMIM (Online Mendelian Inheritance in Man) y en el Orphanet. Se estima que existen alrededor de 10 000 entidades que, según la Organización Mundial de la Salud, afectan al 7% de la población mundial. Estas entidades se pueden clasificar según el tipo de herencia, el tipo de variante genética (por ejemplo, cromosómica o génica) u otros (Tabla 1) (1-3).
Estas entidades genéticas presentan variabilidad clínica o fenotípica, y pueden manifestarse como hipotonía, retraso del desarrollo psicomotor, discapacidad intelectual, epilepsia, neuroregresión, anomalías congénitas, talla corta, microcefalia, inmunodeficiencias primarias, esquizofrenia, trastornos del espectro autista, trastornos de conducta, atención e iperactividad, demencia, movimientos anormales, cáncer; incluso, hay entidades, como la parálisis cerebral infantil, en las que anteriormente no se describía un componente genético y ahora se considera que hasta un 20% de los casos tiene una causa genética (4).
En nuestro país no se ha estimado el número de personas afectadas por enfermedades genéticas; en otras regiones, como en Estados Unidos, se ha estimado que afectan a 13 millones de personas. En ese mismo sentido, representan hasta el 71% de las hospitalizaciones pediátricas, y provocan entre el 20 y el 30% de muertes de este grupo etario (5). Esta proporción de pacientes genera un gran impacto económico en los sistemas de salud; un estudio australiano desarrollado en una cohorte poblacional durante el año 2010 encontró que los pacientes con enfermedades raras generaron el 10,5% de gastos hospitalarios (6), además de una mayor permanencia hospitalaria que sus pares sin condiciones genéticas (6,7).
La mayoría de condiciones genéticas forman parte de las 'enfermedades raras', definidas por la Unión Europea como aquellas con proporciones menores a 1 caso por cada 2 000 personas, estimándose que afectan entre el 6-8% de la población (8).
En el año 2011 se promulgó en Perú la Ley N° 29698: 'Ley que declara de interés nacional y preferente atención el tratamiento de personas que padecen enfermedades raras o huérfanas' (9); no obstante, no se ha generado aún un sistema eficiente de registro de la información sanitaria, ni políticas de inversión científicotecnológicas para el diagnóstico específico de las condiciones genéticas en nuestra población. A pesar de esta falta de registro, se estima que más de dos millones de peruanos tienen una enfermedad genética (10).
La presente publicación tiene como objetivo describir los factores de riesgo frecuentes asociados a condiciones de etiología genética, que incluyen: consanguinidad, antecedentes familiares, edad parental, exposición a teratógenos además de los hábitos y estilos de vida parental. Esta información será útil para los profesionales del sistema de salud, generadores y decisores de políticas públicas.
FACTORES DE RIESGO ASOCIADOS
Antecedentes familiares
En muchas enfermedades genéticas existen antecedentes familiares, los cuales pueden seguir un patrón dominante o recesivo. En las enfermedades con patrón dominante (sean autosómicas o ligadas al cromosoma X) pueden existir antecedentes en los ascendientes. En las entidades recesivas autosómicas es importante buscar el antecedente de hermanos afectados y en las recesivas ligadas al cromosoma X se puede observar ascendientes varones afectados.
Las alteraciones cromosómicas desbalanceadas pueden ser heredadas de padres portadores sanos (con rearreglos balanceados).
Dada la importancia del conocimiento del estado de salud de los ascendientes, una anamnesis acuciosa y un correcto heredograma son una valiosa herramienta en la consulta genética, incluso si las manifestaciones clínicas varían en los familiares afectos. Un heredograma correctamente elaborado representa el 'estetoscopio' del genetista clínico, permite identificar de manera más sencilla, por ejemplo, el patrón de herencia o el número de afectados (11).
Consanguinidad
Actualmente, más de 1,2 billones de personas de la población global tienen un matrimonio consanguíneo, y se estima que el 10,4% de la población mundial está unido con un pariente biológico o es la progenie de una unión consanguínea (12). El coeficiente de endogamia (F) representa la probabilidad de que los dos alelos de un gen en un locus específico sean idénticos y heredados de un ancestro en común (12). La consanguinidad representa la unión entre dos personas descendientes de antecesores comunes, hasta el grado de parentesco de primos de segundo grado o más cercano (F=0,0156).
La prevalencia de matrimonios consanguíneos varía según religión, etnicidad, demografía, residencia (urbana o rural), flujo migratorio, educación, creencias sobre la consanguinidad, entre otros. En nuestro país, no existen datos sobre la prevalencia de consanguinidad; sin embargo, se conoce que existen comunidades con una mayor prevalencia de enfermedades recesivas autosómicas, como la lipodistrofia congénita donde la mayoría afectados proviene de la región de Piura (13-15).
Usualmente el parentesco ancestral es desconocido incluso por los progenitores del paciente, descubriéndose gracias a estudios genéticos como el análisis cromosómico por micromatrices (CMA del inglés Chromosome Microarray Analysis) o el secuenciamiento masivo paralelo (16).
La consanguinidad incrementa el riesgo de autocigocidad (mismos alelos en un locus genético), pudiendo provocar una enfermedad recesiva autosómica con manifestaciones clínicas al nacer, en la infancia o adultez; así como riesgos de abortos recurrentes y enfermedades multifactoriales (17).
De esta manera, la consanguinidad incrementa las probabilidades de que dos progenitores portadores de un alelo alterado tengan un descendiente con una enfermedad genética recesiva autosómica. Esto resalta debido a que en la población general cada persona podría portar 2,8 alelos recesivos autosómicos para enfermedades genéticas pediátricas graves (18) y entre 50 a 100 variantes asociadas a algún trastorno genético (19).
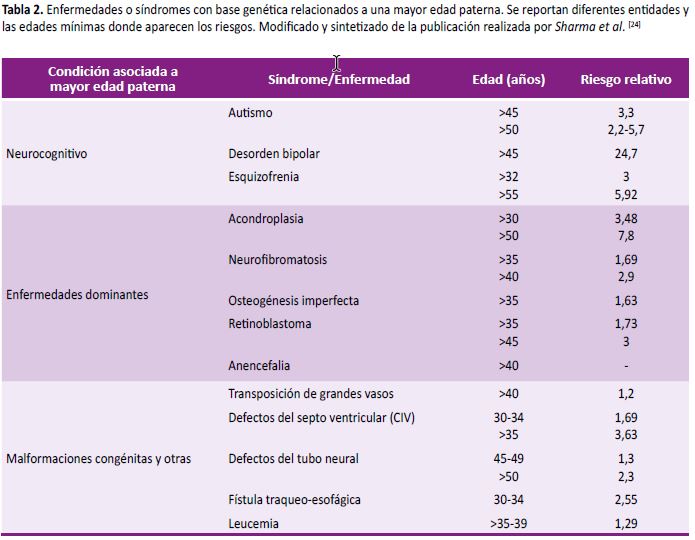
Edad de los padres
A nivel global, la proporción de personas que alcanzan la paternidad a una edad avanzada se ha incrementado. Una investigación mostró que el porcentaje de mujeres que son madres a una edad mayor de 35 años se incrementó de 14,9% en el año 2000 a 19,7% en el 2011 (20). La edad materna avanzada es considerada un factor de riesgo para la presentación de alteraciones cromosómicas numéricas (aneuploidías), en especial trisomías como el síndrome Down, síndrome Patau o síndrome Edwards. La Figura 1a describe la relación entre la edad materna y la frecuencia de concebir descendientes con alteraciones cromosómicas, evidenciándose un aumento exponencial a partir de los 35 años (20).
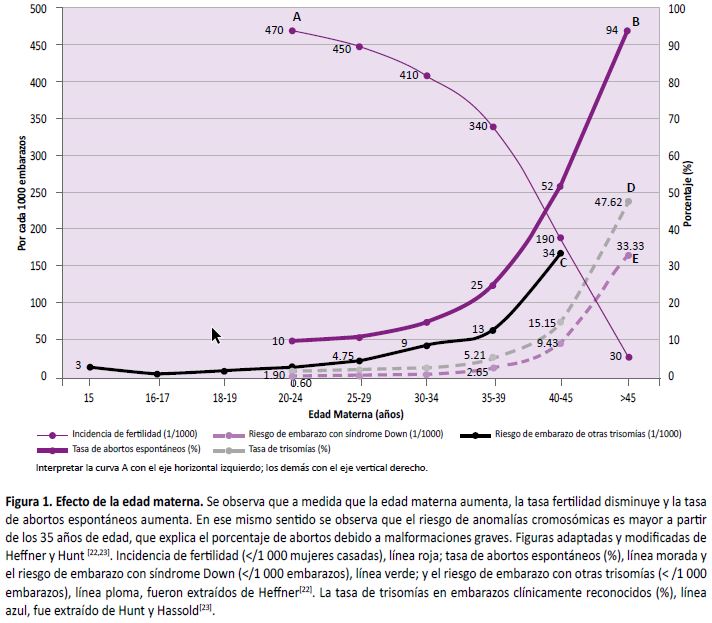
Existe una relación similar entre edad materna con la fertilidad y número de abortos (21) (Figura 1b) (22), debido a fallas en la segregación cromosómica durante la meiosis, mecanismos que están directamente relacionados con la edad materna, conllevando a una pérdida prematura de la cohesión centromérica y disfunción proteica en la homeostasis de la segregación cromosómica meiótica (20,23).
En el caso de los varones, el envejecimiento afecta a procesos relacionados con daño al ADN, acortamiento telomérico, senescencia o apoptosis; lo que conlleva a la acumulación de mutaciones de novo para enfermedades genéticas (por ejemplo: síndrome de Noonan, síndrome de Marfan, acondroplasia, etc.), malformaciones congénitas, autismo, cáncer, esquizofrenia, entre otros; donde dependiendo de la edad del padre, existe cierto riesgo para la presentación de dichas entidades, el cual se encuentra por encima de los 30 años de edad (23). De otro lado, el envejecimiento es factor de riesgo para la aparición de aneuploidías; un estudio desarrollado en padres de entre 45 y 49 años demostró que había un riesgo 3 veces mayor de tener hijos con trisomía 21 comparado con padres jóvenes (24).
Uso de técnicas reproductivas in vitro
Las técnicas reproductivas in vitro son usadas cada vez más debido a, entre otras razones, la postergación de la maternidad, así como la infertilidad de la pareja. En el año 1991, un estudio realizado mediante cariotipo convencional a 11 615 espermatozoides y 772 oocitos de personas sanas mostró alteraciones cromosómicas en el 3% y 19%, respectivamente (26).
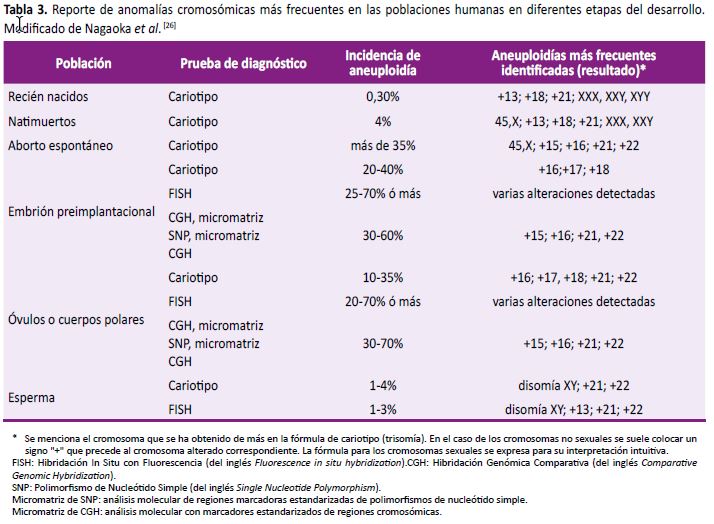
Cuando se utilizan técnicas como hibridación in situ con fluorescencia (FISH) y análisis cromosómico por micromatrices (CMA), el porcentaje de detección de alteraciones cromosómicas puede llegar al 70% (27). En la Tabla 3, se muestran los datos recopilados por Nagaoka et al. en su estudio para la detección de aneuploidías en grupos poblacionales mediante diferentes técnicas de diagnóstico genético y molecular (27); no obstante, estos hallazgos podrían estar per se relacionados con una edad parental avanzada o por la condición de infertilidad –y no al uso de técnicas reproductivas in vitro-, que están asociados con: falla meiótica del ovocito, alteraciones hormonales, errores en la reparación del ADN, provocando con ello diversas condiciones genéticas (27). En el Perú no hay una reglamentación que regule la práctica de la reproducción asistida.
Teratógenos
Los teratógenos son insultos ambientales (por ejemplo: agente mecánico, hipoxia, deficiencia de nutrientes, infecciones intrauterinas, etc.) que durante la gestación provocan alteraciones permanentes (estructurales o funcionales) en el desarrollo (28).
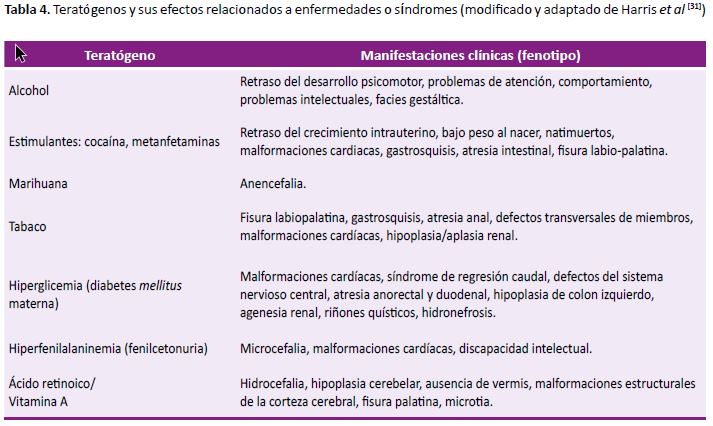
Por ejemplo, el misoprostol, un análogo semisintético de prostaglandina E1 utilizado para la prevención y tratamiento de las úlceras gástricas y duodenales, es un medicamento teratógeno y abortivo. Se ha reportado su uso en el Perú, con una venta indiscriminada sin prescripción médica, inclusive desde páginas de internet (29). Aun cuando luego de su uso no se concluya en aborto, existe el riesgo de disrupción en el desarrollo embrionario, como el síndrome Moebius, defectos en el desarrollo de las extremidades, malformaciones cerebrales, gastrosquisis, artrogriposis e hidrocefalia (30). En la Tabla 4 se muestra una lista de algunos teratógenos conocidos y sus efectos asociados (31).
Otros factores asociados
La obesidad, el consumo de una dieta de baja calidad y gestaciones múltiples están asociados a defectos del tubo neural, fisuras orofaciales, hipoplasia cardiaca izquierda, estenosis pulmonar, tetralogía de Fallot, hernia diafragmática, irenomelia, holoprosencefalia, atresia intestinal, aplasia cutis, defectos transversos de extremidades, entre otras malformaciones (31).
El consumo de cocaína puede provocar defectos epigenéticos en el ADN espermático, con posibles efectos deletéreos del comportamiento y aprendizaje en la descendencia (32,33). Los padres que consumen alcohol de manera sostenida y en cantidades nocivas tendrían mayor probabilidad de tener hijos con trastornos psicosociales, discapacidad intelectual, cambios en la personalidad, desorden de hiperactividad y déficit de atención, entre otros trastornos psiquiátricos además de riesgo elevado de cáncer pediátrico (33).
DISCUSIÓN
La incidencia de las enfermedades o síndromes genéticos evaluada aisladamente no generaría aparentemente un impacto en la salud pública; sin embargo, si la analizamos en su totalidad constituye una carga importante para los sistemas sanitarios, más en los países que están en vías de desarrollo.
En la Unión Europea, se estimó que 6 a 8% de la población tendría alguna enfermedad rara y que el 80% de este porcentaje sería de etiología genética; además, la mitad de estas enfermedades genéticas se presentaría como síndromes en la etapa pediátrica (por ejemplo: neurofibromatosis, acondroplasia, retraso del desarrollo o discapacidad intelectual, entre otros), mientras que la otra mitad se manifestaría en la etapa adulta (por ejemplo: enfermedad Huntington, Crohn, esclerosis lateral amiotrófica, cáncer de tiroides, entre otras) (35). Si consideramos la proporción estimada en la Unión Europea (6,4% tendría enfermedades de etiología genética), cerca de 2 millones de peruanos presentaría una enfermedad genética, de los cuales la mayoría no ha tenido una evaluación y diagnóstico precisos.
La identificación sistemática, dentro de un sistema de salud, de los factores de riesgo de estas condiciones de salud representaría una alternativa de prevención; por ejemplo, identificando oportunamente enfermedades genéticas recesivas como la atrofia muscular espinal o la distrofia muscular de Duchenne, lo cual abriría la posibilidad de ofrecer un asesoramiento genético exacto, así como una terapia personalizada (por ejemplo: oligonucleótidos sintéticos).
Por lo tanto, existe la necesidad de crear e implementar servicios de Genética Médica y Medicina Genómica, acordes a los estándares internacionales, que permitirán que la población tenga acceso a una medicina de precisión, llámese a aquella que utiliza la información sobre los genes o proteínas de una persona con el fin de determinar el diagnóstico y por consiguiente una terapéutica específica.
Además, es importante una mayor difusión en el pregrado de las facultades de medicina y relacionados sobre el reconocimiento oportuno de las enfermedades genéticas (36).
El trabajo multidisciplinario fortalecerá la detección, manejo y prevención de estas condiciones, por lo que es necesario impulsar la organización y facilitar el desenvolvimiento de estos equipos. La educación de los pacientes y actores sociales; la interacción con asociaciones de pacientes, el diálogo dinámico, recíproco y vinculante entre profesionales de salud, administradores, gestores y decisores en políticas públicas, son estrategias para el manejo de equipos multi e interdisciplinarios (36,37).
Es difícil realizar estudios costo-beneficio en pacientes con enfermedades raras, debido a su baja incidencia y prevalencia cuando se analizan por separado. No obstante, se han desarrollado estudios económicos para el uso de CMA, primer test de elección en el diagnóstico de discapacidad intelectual, demostrando ser costo-efectivo en relación a otros test convencionales como el cariotipo (38).
El desarrollo de actividades que incentiven la participación de los pacientes, así como la adecuada organización/planificación en los gastos del sistema de salud, podrían beneficiar a la sociedad siguiendo los principios de justicia y no maleficencia. Las tecnologías desarrolladas para el diagnóstico y tratamiento de personas con enfermedades raras avanzan vertiginosamente y demuestran también utilidad clínica para enfermedades comunes (39).
Nuestro país requiere estrategias que integren esfuerzos para incentivar el interés en la investigación de estas condiciones genéticas. Además, evitar la discriminación mediante la implementación de tecnologías diagnósticas en los hospitales que cuentan con estos servicios, así como en todas las regiones de nuestro territorio, lo cual permitirá un diagnóstico oportuno, acceso a terapias específicas, asesoramiento genético integral, permitirá establecer políticas de salud pública que disminuyan la incidencia o prevalencia, así como los costos en diagnóstico y tratamiento.
CONCLUSIÓN
La identificación de factores de riesgo representa una estrategia preventiva de enfermedades genéticas. Por ejemplo, el reconocimiento masivo que la edad parental avanzada o la consanguinidad son factores de riesgo en la aparición de enfermedades genéticas provocarían que la población tome decisiones informadas respecto a su paternidad.
Es necesario implementar un sistema multi e interdisciplinario para el diagnóstico precoz y oportuno pre y posconcepcional; así como al acceso de tratamientos tempranos y específicos, en concordancia a los estándares aplicados en otros países. Es importante educar y capacitar en Genética a los profesionales de salud y población general, con el fin de empoderar a los actores sociales y políticos, de acuerdo al conocimiento científico y la implementación biotecnológica en este campo; lo que reivindicaría a este grupo de pacientes que se encuentran relegados actualmente por nuestro sistema de salud.
REFERENCIAS BIBLIOGRÁFICAS
1. Human Genetics Knowledge – OMIM. Online Mendelian Inheritance in Man (Internet). Baltimore: OMIN; c1966-2018 (citado 14 de julio de 2017). Disponible en: https://www.omim.org/ [ Links ]
2. World Health Organization. Genes and human disease (Internet). Geneva: WHO; c2018 (citado 14 de julio de 2017). Disponible en: http://www.who.int/genomics/public/geneticdiseases/en/index2.html [ Links ]
3. Cortés F. Las enfermedades raras. Rev Med Clin Condes. 2015;26(4):425-31. [ Links ]
4. Fahey MC, Maclennan AH, Kretzschmar D, Gecz J, Kruer MC. The genetic basis of cerebral palsy. Dev Med Child Neurol. 2017;59(5):462-9. [ Links ]
5. Kingsmore S. Comprehensive carrier screening and molecular diagnostic testing for recessive childhood diseases. PLoS Curr. 2012:e4f9877ab8ffa9. doi: 10.1371/4f9877ab8ffa9. [ Links ]
6. Walker CE, Mahede T, Davis G, Miller LJ, Girschik J, Brameld K, et al. The collective impact of rare diseases in Western Australia: an estimate using a population-based cohort. Genet Med. 2017;19(5):546-52. [ Links ]
7. McCandless SE, Brunger JW, Cassidy SB. The Burden of Genetic Disease on Inpatient Care in a Children’s Hospital. Am J Hum Genet. 2004;74(1):121-7.
8. Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999 on orphan medicinal products (Internet). Brussels: European Commission: 2000 (citado 10 de marzo de 2017). Disponible en: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2000_141_cons-2009-07/reg_2000_141_cons-2009-07_en.pdf [ Links ]
9. Ley que declara de interés nacional y preferente atención el tratamiento de personas que padecen enfermedades raras o huérfanas. El Peruano. 2011, Ley N° 29698 p. 443816 (26 de mayo de 2011). Disponible en: http://www.minsa.gob.pe/erh/documentos/Ley_29698.pdf [ Links ]
10. Ministerio de Salud. Pre-publicación del proyecto de Plan nacional de Prevención, Diagnóstico, Atención Integral, tratamiento, Rehabilitación, y Monitoreo de las Enfermedades Raras o Huérfanas. 2014. RM N° 198-2014/MINSA. (Internet). Lima: Minsa; 2014 (citado 18 de julio de 2017). Disponible en: http://www.minsa.gob.pe/erh/documentos/RM198_2014_MINSA.pdf [ Links ]
11. Rimoin DL, Pyeritz RE, Korf B. Emery and Rimoin’s principles and practice of medical genetics. 6th ed. San Diego, California: Academic Press; 2013.
12. Fareed M, Afzal M. Genetics of consanguinity and inbreeding in health and disease. Ann Hum Biol. 2017;44(2):99-107. [ Links ]
13. Bittles AH, Black ML. Evolution in health and medicine Sackler colloquium: Consanguinity, human evolution, and complex diseases. Proc Natl Acad Sci U S A. 2010;107 Suppl 1:1779-86. [ Links ]
14. Liascovich R, Rittler M, Castilla EE. Consanguinity in South America: Demographic Aspects. Hum Hered. 2000;51(1-2):27-34. [ Links ]
15. Carr IM, Bhaskar S, O’ Sullivan J, Aldahmesh MA, Shamseldin HE, Markham AF, et al. Autozygosity Mapping with Exome Sequence Data. Hum Mutat. 2013;34(1):50-6.
16. Purizaca-Rosillo N, Mori T, Benites-Cóndor Y, Hisama FM, Martin GM, Oshima J. High incidence of BSCL2 intragenic recombinational mutation in Peruvian type 2 Berardinelli-Seip syndrome. Am J Med Genet A. 2017;173(2):471-478. [ Links ]
17. Zaghlol RY, Haghighi A, Alkhayyat MM, Theyab OF, Owaydah AM, Massad MM, et al. Consanguinity and the Risk of Hashimoto’s Thyroiditis. Thyroid off J Am Thyroid Assoc. 2017;27(3):390-5.
18. Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, et al. Carrier Testing for Severe Childhood Recessive Diseases by Next-Generation Sequencing. Sci Transl Med. 2011;3(65):65ra4. [ Links ]
19. 1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061-73. [ Links ]
20. Jacobs M, Cooper S-A, McGowan R, Nelson SM, Pell JP. Pregnancy outcome following prenatal diagnosis of chromosomal anomaly: a record linkage study of 26,261 pregnancies. PLoS ONE. 2016;11(12):e0166909. [ Links ]
21. Hook EB, Cross PK, Schreinemachers DM. Chromosomal Abnormality Rates at Amniocentesis and in Live-Born Infants. JAMA. 1983;249(15):2034-8. [ Links ]
22. Heffner LJ. Advanced Maternal Age — How Old Is Too Old? N Engl J Med. 2004;351(19):1927-9. [ Links ]
23. Hunt P, Hassold T. Female meiosis: coming unglued with age. Curr Biol. 2010;20(17):R699-702. [ Links ]
24. Sharma R, Agarwal A, Rohra VK, Assidi M, Abu-Elmagd M, Turki RF. Effects of increased paternal age on sperm quality, reproductive outcome and associated epigenetic risks to offspring. Reprod Biol Endocrinol RBE. 2015;13:15. [ Links ]
25. Conti SL, Eisenberg ML. Paternal aging and increased risk of congenital disease, psychiatric disorders, and cancer. Asian J Androl. 2016;18(3):420-4. [ Links ]
26. Martin RH, Ko E, Rademaker A. Distribution of aneuploidy in human gametes: comparison between human sperm and oocytes. Am J Med Genet. 1991 Jun 1;39(3):321-31. [ Links ]
27. Nagaoka SI, Hassold TJ, Hunt PA. Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet. 2012;13(7):493-504. [ Links ]
28. Klein E, Gallardo B, Chávez M, Abarca-Barriga H. Atlas de dismorfología pediátrica. 1o Edición. Lima: Instituto Nacional de Salud del Niño; 2012. [ Links ]
29. Peligroso abortivo se vende ilegalmente hasta por Facebook (Internet). El Comercio. 2014 (citado 12 de marzo de 2017). Disponible en: https://elcomercio.pe/lima/peligroso-abortivo-vende-ilegalmente-facebook-330570 [ Links ]
30. Cavieres MF. Toxicidad del misoprostol sobre la gestación. Revisión de la literatura. Rev Méd Chile. 2011;139(4):516-23. [ Links ]
31. Harris BS, Bishop KC, Kemeny HR, Walker JS, Rhee E, Kuller JA. Risk factors for birth defects. Obstet Gynecol Surv. 2017;72(2):123-35. [ Links ]
32. He F, Lidow IA, Lidow MS. Consequences of paternal cocaine exposure in mice. Neurotoxicol Teratol. 2006;28(2):198-209. [ Links ]
33. Wimmer ME, Briand LA, Fant B, Guercio LA, Arreola AC, Schmidt HD, et al. Paternal cocaine taking elicits epigenetic remodeling and memory deficits in male progeny. Mol Psychiatry. 2017;22(11):1653. [ Links ]
34. Finegersh A, Rompala GR, Martin DIK, Homanics GE. Drinking beyond a lifetime: New and emerging insights into paternal alcohol exposure on subsequent generations. Alcohol. 2015;49(5):461-70. [ Links ]
35. Orphanet. About rare diseases (Internet). Paris, France: Orphanet;2012 (citado 14 de julio de 2017). Disponible en: https://www.orpha.net/consor/cgi-bin/Education_AboutRareDiseases.php?lng=EN [ Links ]
36. World Health Organization. About WHO’s Human Genomics in Global Health initiative (Internet). Geneva: WHO; c2018 (citado 13 de marzo de 2017). Disponible en: http://www.who.int/genomics/about/en/
37. Hamilton AB, Oishi S, Yano EM, Gammage CE, Marshall NJ, Scheuner MT. Factors influencing organizational adoption and implementation of clinical genetic services. Genet Med. 2014;16(3):238-45. [ Links ]
38. Trakadis Y1, Shevell M. Microarray as a first genetic test in global developmental delay: a cost-effectiveness analysis Dev Med Child Neurol. 2011 Nov;53(11):994-9. [ Links ]
39. Lu CY, Cohen JP. Can genomic medicine improve financial sustainability of health systems? Mol Diagn Ther. 2015;19(2):71-7. [ Links ]
Conflictos de Interés: Los autores declaran no tener conflictos de interés.
Fuentes de Financiamiento: Autofinanciado.
Correspondencia
Hugo H. Abarca Barriga habarca@insn.gob.pe
Recibido: 14/09/2017
Aprobado: 28/02/2018

