











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Los trastornos del neurodesarrollo son entidades que alteran el desarrollo del sistema nervioso central. Esto implica la disfunción del desarrollo cerebral, que puede manifestarse como problemas neuropsiquiátricos, del aprendizaje, el lenguaje o comunicación no verbal y funciones motoras. Se incluyen, entre otros, a la discapacidad intelectual (DI), retraso del desarrollo psicomotor (RDPM) y trastorno del espectro autista (TEA) 1.
El RDPM representa la no adquisición normal de al menos dos habilidades psicomotrices, debido a que el niño no alcanzó alguno de los hitos del desarrollo, los alcanzó después del tiempo esperado o de una manera incompleta 2. La DI se define como la limitación en dos áreas: la inteligencia o capacidad mental y el comportamiento adaptativo en cualquiera de sus tres dominios: conceptual, social y práctico 3. Los trastornos del espectro autista, constituyen un grupo complejo de entidades que afectan el neurodesarrollo que se presenta en la niñez. Mientras que las personas con TEA se caracterizan por presentar deficiencia persistente en la comunicación social y la interacción social en múltiples contextos, así como la presencia de patrones de comportamiento, intereses o actividades restringidas y repetitivas 4.
Por otro lado, la talla baja idiopática (TBI) es definida cuando ésta se encuentra por debajo de dos desviaciones estándar sin evidencia de alguna patología 5. Las anomalías congénitas son condiciones de origen prenatal que están presentes al nacimiento y que potencialmente impactan en el desarrollo, salud y sobrevida. Éstas se denominan sindrómicas cuando presentan un patrón reconocible y con una causa probable (genética o teratogénica) 6.
La prevalencia mundial de la DI oscila entre el 1 y 3% de la población, teniendo una etiología genética que podría llegar hasta el 90% de los casos, dependiendo del uso combinado del análisis cromosómico por micromatrices y el secuenciamiento exómico/genómico 7-9. En Latinoamérica, la prevalencia de DI es de 3,0-12,9% de la población 10, estimándose que en el Perú tendrían DI entre 0,9 y 3,8 millones de personas. La prevalencia del TEA en menores de 18 años de edad es de 1,5-2% (3,11 ). Mientras la prevalencia de talla baja en niños y de recién nacidos con anomalías congénitas es del 4,6% 12 y 1-3%, respectivamente 13. Durante el 2015-2018, en el sevicio de Genética y errores innatos del desarrollo (Genética & EIM) del Instituto Nacional de Salud del Niño (INSN) se evaluaron a 10 524 niños, de los cuales el 19% tenía un trastorno del neurodesarrollo (RDPM, DI, TEA), 3,79% talla baja y el 1,06% síndrome malformativo 14.
El análisis cromosómico por micromatrices o CMA (del inglés Chromosomal Microarray Analysis) permite detectar variantes en el número de copias o CNVs (del inglés Copy Number Variation) las cuales se definen como segmentos en ganancia o pérdida de ADN iguales o mayores a 1 Kpb. Las CNVs se clasifican en cinco tipos según su patogenicidad en : i) CNV patogénica; ii) CNV probablemente patogénica; iii) CNV de significado incierto); iv) CNV probablemente benigna y v) CNV benigna 15,16. Las CNVs pueden provocar alteración en la expresión génica mediante el fenómeno de dosis génica, fusión de genes, disrupción de genes o la alteración en los efectos regulatorios, los cuales provocan modificación de vías importantes y, dependiendo de la región genómica comprometida, causarán un fenotipo determinado 17. Además, el CMA permite, también, ubicar regiones en homocigosidad o ROH (del inglés Regions Of Homozygosity), que nos muestra las disomías uniparentales cuando el ROH es mayor a 10 Mpb, y la consanguinidad parental si el total de ROHs de las regiones autosómicas es mayor a 2,56% 18-20. Las disomías uniparentales podrían provocar según la región comprometida alteraciones en genes susceptibles de impronta o la aparición de una enfermedad recesiva autosómica 20.
Las CNVs son la etiología del TEA y DI entre el 20% y 30% de los casos, respectivamente, mientras que el uso del secuenciamiento masivo puede facilitar el diagnóstico hasta en el 68,3% 8,10,21,22.
Con relación a talla baja idiopática el CMA puede detectar la etiología en el 2,5% 23.
Desde el año 2010, el CMA está indicada como la prueba diagnóstica de primera línea en pacientes con RDPM, DI, TEA, TBI y síndrome malformativo; sin embargo, desde el 2019 el secuenciamiento exómico o genómico está considerado como la primera prueba diagnóstica 9,18,21,24-29. El objetivo del estudio fue determinar las CNVs, ROH superiores al 2,56%, y ROH segmentales de más de 10 Mpb mediante el uso de CMA en niños peruanos con trastornos del neurodesarrollo, síndrome malformativo y talla baja idiopática.
MATERIALES Y MÉTODOS
Diseño y tipo de estudio
Es un estudio tipo descriptivo y transversal donde se revisaron los datos consignados en las historias clínicas de todos los pacientes entre 0-18 años que acudieron al Servicio de Genética & EIM del INSN entre diciembre de 2015 a enero de 2018.
Población y muestra
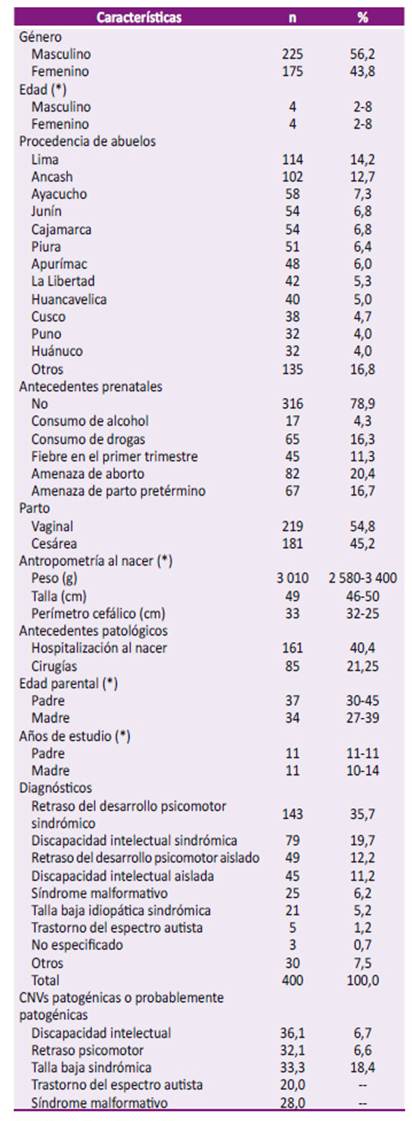
Se realizó el CMA a 400 pacientes, analizando los resultados de todos aquellos que presentaban retraso del desarrollo psicomotor, discapacidad intelectual, trastorno del espectro autista, talla baja idiopática y/o síndrome malformativo (n=367). Se subclasificó en entidades aisladas o sindrómicas dependiendo de la presencia de comorbilidades, es así que, los pacientes con DI y RDPM se clasificaron como sindrómicos si tenían presencia de TEA, alteraciones de la antropometría o dismorfias; y la talla baja se definió como sindrómica si se asociaba a otros cambios antropométricos u a otras anomalías congénitas menores (Tabla 1). Se excluyó del CMA a los pacientes que clínicamente padecían de una enfermedad monogénica y que la probabilidad de la alteración fuera una variante de un único nucleótido o de múltiples nucléotidos.
Variables de estudio
Las CNVs fueron comparadas con las bases de datos genómicas: Database of Chromosomal Imbalance, Phenotype in Humans using Ensemble Resources (DECIPHER), y de University of California, Santa Cruz (UCSC). Las CNVs encontrados se clasificaron según la ACMG (American College of Medical Genetics) 15,16. Los resultados del CMA se clasificaron como resultados anormales si el análisis demostraba CNVs patogénicas o probabalemente patogénicas; o aquellos que presentaban al menos una región con un ROH mayor a 10 Mpb y/o si el total de ROHs de las regiones autosómicas era mayor a 2,56% 20.
Procedimiento
El CMA se realizó con una muestra de sangre periférica, de la cual se extrajo el ADN genómico (250ng); el cual fue amplificado, etiquetado e hibridado usando el protocolo GeneChip CytoScan 750K Array (Affymetrix, USA ®) de acuerdo con las instrucciones del fabricante. La prueba incluye 550 000 marcadores no polimórficos y 200 436 marcadores SNP (del inglés single nucleotide polymorphism). Las celdas en filas fueron escaneadas mediante el programa informático Chromosome Analysis Suite (ChAS) (Affymetrix, USA ®). Las ganancias o pérdidas son consideradas cuando se comprometen al menos 50/25 marcadores respectivamente. Las ROH comprometen una longitud de al menos 10 Mpb (ver Thermo Fisher Sc Inc, 2017). A todos los padres se les brindó el asesoramiento genético correspondiente y se realizó seguimiento de manera ambulatoria.
Análisis estadístico
El análisis descriptivo se realizó mediante el uso de frecuencias y porcentajes. Para determinar si existió asociación entre el tipo de diagnóstico y la presencia o ausencia de CNVs, se usó la prueba de chi cuadrado. Además, se determinó la concordancia entre el CMA y el cariotipo, así como los valores predictivos positivos y negativos, sensibilidad y especificidad de estas pruebas.
Aspectos éticos
Se obtuvo la aprobación del comité de Ética del INSN, preservando la privacidad y confidencialidad de los datos clínicos y genómicos de los sujetos de investigación y sus respectivas familias, de acuerdo con los lineamientos de las buenas prácticas clínicas y de ética en investigación biomédica.
RESULTADOS
El 56,3% (n=225) fueron varones y el 43,8% (n=175) mujeres, con un promedio de la edad de los niños atendidos de 5,6 ± 4,8 años. Otras variables consideradas en el estudio fueron: antecedentes prenatales y natales, procedencia de abuelos, edad parental y los diagnósticos por lo que se indicó el CMA (Tabla 1). Con respecto a la escolaridad, el 23,9% acudió a un centro educativo básico alternativo. El promedio del coeficiente intelectual según la escala de inteligencia Stanford-Binet en los pacientes con DI fue de 58,1 ± 25,3 puntos.
En el 82,1% de los casos tenían al menos una prueba complementaria, las cuales en su mayoría fueron no concluyentes. Estas pruebas fueron: cariotipo en sangre periférica (39,6%), RMN de encéfalo (31,3%), TEM de encéfalo (30,4%), electroencefalograma (24,6%), potenciales evocados auditivos (22,9%), espectrometría de masas en tándem (5,5%) y fragilidad cromosómica (1,3%). Los hallazgos antropométricos alterados fueron talla baja (34,6%), microcefalia (27,1%), bajo peso (17,9%), obesidad (6,3%), macrocefalia (5,4%), sobrepeso (5,0%) y talla alta (3,8%).
En el 49,9% de todos los casos analizados (n=367), se evidenció un resultado normal de CMA. Por otro lado, en el 26,0% de los pacientes se encontró una o más CNVs patogénicas o probablemente patogénicas; en el 15,7% se encontró una o más de CNVs patogénicas o probablemente patogénicas más ROH superior a 10 Mb.
Además, se encontró un ROH mayor a 2,56% asociado o no a CNVs patogénicas, en el 6,2% de los casos. Por otro lado, se evidenciaron CNVs probablemente patogénicas en el 29,5%; y CNVs de significado incierto en combinación con ROH en el 6,8%. En el 19,6% (n=72) de los pacientes se planteó una posible disomía uniparental, de los cuales en el 56,7% de los casos, esta fue observada en el cromosoma X, y en el resto de los niños, en uno o más cromosomas.
Con relación a los casos clasificados como discapacidad intelectual no sindrómica, se observó que en el 51,1% de los pacientes mostró un resultado anormal. En los pacientes clasificados como discapacidad intelectual sindrómica los resultados anormales se observaron en el 54,5%, sin observarse diferencias significativas entre estos grupos (p=0,770). En este grupo, se observó siete pacientes, como característica adicional, al TEA. Adicionalmente, dos de ellos presentaron CNVs patogénicas.
En los pacientes con retraso del desarrollo psicomotor aislado o sindrómico, se observó que el 49,0% y 47,6%, respectivamente presentaron resultados anormales en el CMA. Se observaron ocho pacientes con RDPM sindrómico que también presentaban TEA, de los cuales uno tenía ROH de 3,0%. El CMA mostró resultados anormales en un 20% de los niños con diagnóstico del trastorno de espectro autista y en el 33,3% de los pacientes con talla baja idiopática sindrómica (Figura 1). En aquellos pacientes que tuvieron el diagnóstico de un síndrome malformativo no especificado (n=25), se pudo apreciar que el 52% de los niños tuvieron un CMA anormal. Mientras que, en el total de pacientes evaluados, en 95 casos se encontraron CNVs patogénicas, de los cuales, 20 niños presentaron más de una CNV.
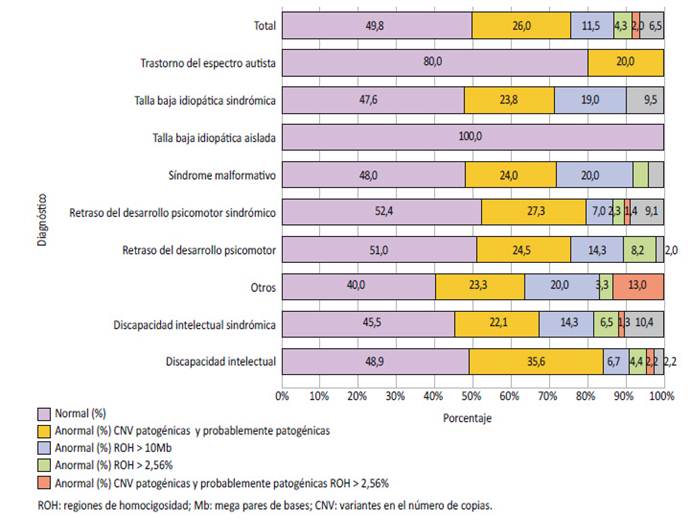
Figura 1 Distribución según los diagnósticos y los resultados de los análisis cromosómicos por micromatrices en pacientes con trastornos del neurodesarrollo, talla baja y síndrome malformativo.
El tamaño de las CNVs patogénicas varían entre 10,3 kb y 155 065 kb, con un promedio de 15 689,7 kb. El número total de CNVs patogénicas encontradas fueron de 117, de los cuales 74 fueron en pérdida, 42 en ganancia y cinco, en mosaico (tres en ganancia y dos en pérdida) (Figura 2). En 54 niños se observaron CNVs probablemente patogénicas, de los cuales 45 tuvieron CNV en ganancia y nueve CNVs en pérdida (Figura 3 y material suplementario 1).
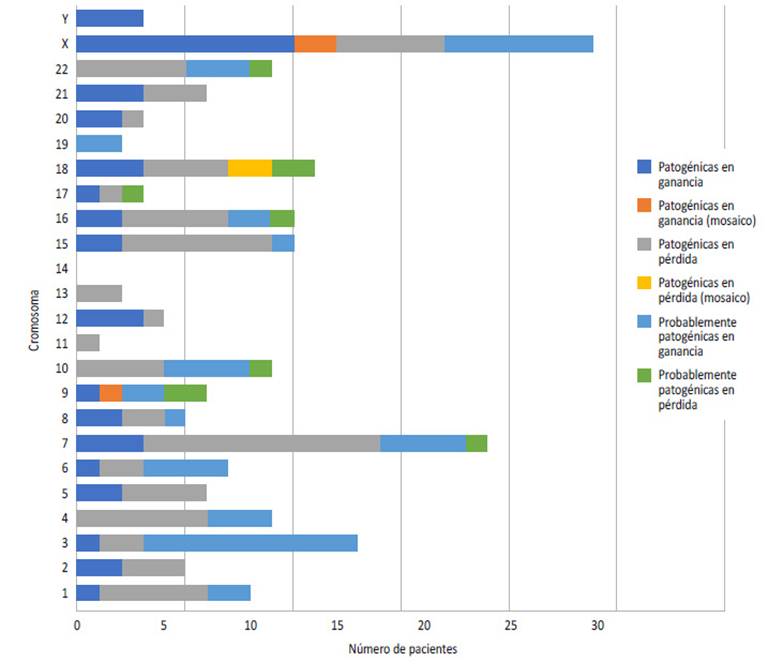
Figura 2 Frecuencia de aparición de variantes en el número de copias patogénicas o probablemente patogénicas según ubicación cromosómica.
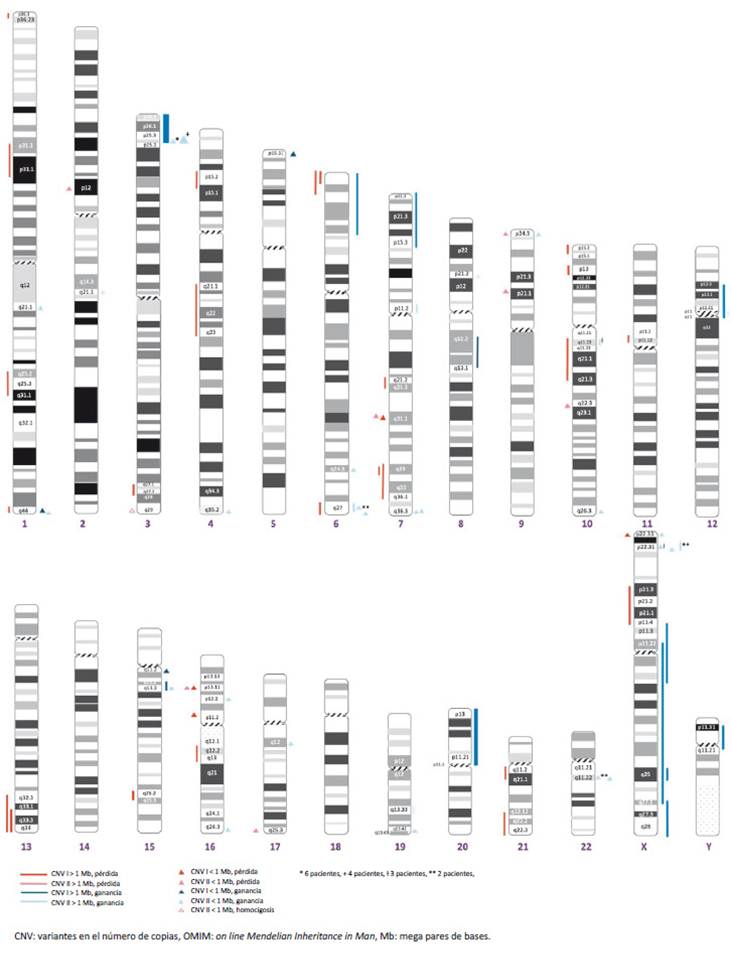
Figura 3 Ideograma con las CNVs patogénicas o probablemente patogénicas. Se ha excluido a CNVs que están registrados en el OMIM y aquellos como producto de un cromosoma derivado (material suplementario 1). Las líneas y los triángulos más gruesos y largos representan a triplicaciones.
En cuatro pacientes con diagnósticos de RDPM sindrómico y DI sindrómica, se identificó un cromosoma marcador. En dos de ellos se pudo detectar el origen del material genético extra, a través del CMA en el cromosoma X y en el cromosoma 7; mientras que, en los otros dos el CMA fue normal. En tres niños con diagnóstico de retraso del desarrollo psicomotor (n=2) y discapacidad intelectual (n=1) se encontró material adicional (add) en el cromosoma 8, 15 y 13 respectivamente. Al realizarles el CMA se encontró CNV en los cromosomas 7 y 8, 15 y 16, y 20 respectivamente.
La concordancia entre el cariotipo y el CMA fue bajo (valor kappa =0,62). En 21 pacientes no hubo relación entre los resultados del análisis cromosómico por micromatrices y el cariotipo en sangre periférica, mostrando que en dos de los casos en los que el cariotipo se realizó a priori, se tenía un cromosoma marcador; sin embargo, en el CMA no se pudo detectar el origen del material genético extra. En los otros 19 pacientes, quienes también tenían un estudio citogenético convencional normal previo, éstos acudieron al servicio para realizar el CMA en el que se pudo detectar CNVs patogénicas por encima de la resolución de un cariotipo convencional. En 22 niños se encontró una relación de los resultados entre el cariotipo y el CMA hallando una CNV patogénica; además, en 186 casos tuvieron un resultado normal en los que hubo relación entre el cariotipo y el CMA. Los valores de sensibilidad y especificidad fueron de 53,7% y 98,9% respectivamente. Mientras que los resultados del valor predictivo positivo y negativo (asumiendo un 1% de prevalencia en la población), están en el 33,7% y 99,5% respectivamente.
DISCUSIÓN
Sabemos que la etiología de la discapacidad intelectual, retraso del desarrollo psicomotor, síndromes malformativos y trastorno del espectro autista es genética. En este estudio se observaron más CNVs patogénicas y probablemente patogénicas en pacientes con DI (36,1%) en comparación a lo reportado en la literatura que es alrededor del 15-28% 22,25,30; sin embargo, en pacientes con DI y dismorfia facial el rango puede llegar hasta en el 33,3% 31. En ese mismo sentido, se identificaron más CNVs patogénicas o probablemente patogénicas en los pacientes con retraso psicomotor aislado o sindrómico (32,14% vs 15-22%). Al comparar el hallazgo de las CNVs que ocasionan talla baja idiopática sindrómica se encontró una frecuencia mayor a los reportado (33,3% vs 2,5%) 23; mostrando que estos porcentajes se encuentran por encima de lo reportado previamente.
Por otro lado, no se encontró diferencias en los pacientes diagnosticados con TEA ya que coincidió con lo reportado de las CNVs en otros estudios previos (20%). Estas diferencias de mayor tasa de detección son esperadas, debido a que en muchos países los médicos solicitan el CMA como primer test para el diagnóstico 32; mientras que en nuestra institución es solicitada exclusivamente por los médicos genetistas, quienes tendrían mayor posibilidad de diferenciar y excluir condiciones monogénicas o teratogénicas.
En los pacientes que presentaban RDPM y DI con CNVs de significado incierto, se concluyó que por la frecuencia aumentada de estas variantes y su tamaño menor a 500 kpb, es probable que muchas de ellas estarían siendo reclasificadas en probablemente benignas o benignas 16. Sin embargo, es importante precisar que cuando el tamaño es mayor a 500 kpb podría estar a favor de que la CNV sea interpretado como probablemente patogénico observando nueve CNVs no reportados previamente (Tabla 2)33.
Tabla 2 Variantes en el número de copias de significado incierto raras que podrían ser clasificadas como probablemente patogénicas.
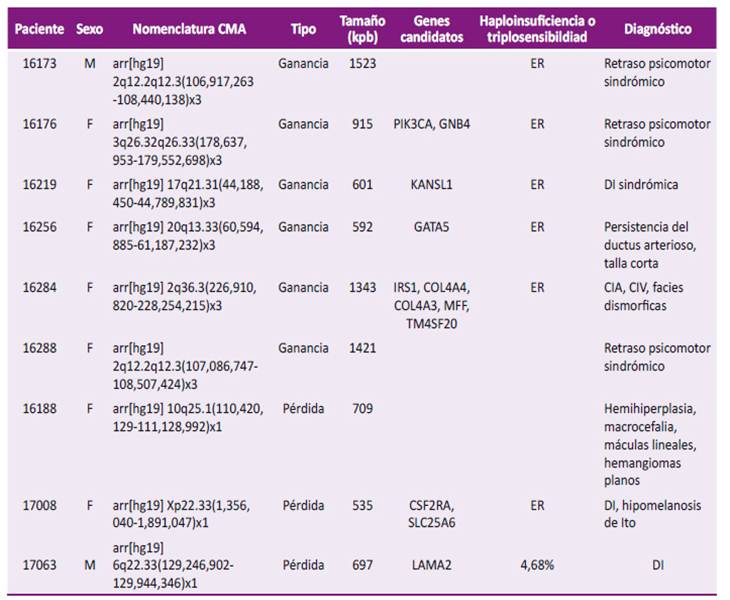
Kpb: kilopares de bases; ER: en revisión; DI: discapacidad intelectual; CIA: comunicación interauricular; CIV: comunicación interventricular; F: sexo femenino; M: sexo masculino.
En el 6,2% de los pacientes se observó ROH por encima de 68 Mb de los cromosomas autosómicos. Esto significa, que los padres de los niños evaluados eran consanguíneos con un grado de parentesco correspondiente hasta de cuarto grado. Esta proporción de pacientes con consanguinidad parental no declarada, se encuentra por encima de lo descrito en otras regiones, como en Brasil, con una frecuencia de 3,2% de los casos 19. En este mismo sentido, hemos observado de manera muy preocupante que el 2% de los padres de los pacientes eran consanguíneos de primer grado no declarado previamente (Tabla 3), lo cual también se encuentra por encima de lo reportado previamente (0,5%) 19.
Tabla 3 Tamaño de ROH y coeficiente de endogamia en los pacientes evaluados en el INSN.
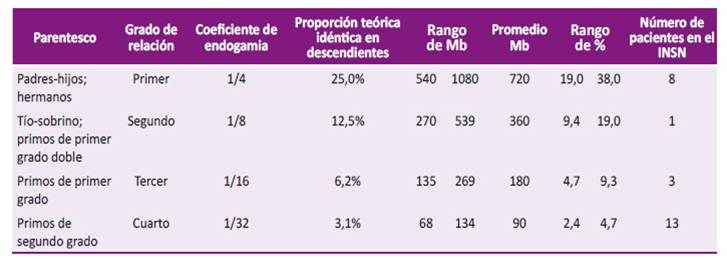
ROH: regiones de homocigosidad; Mb: megapares de base; INSN: Instituto Nacional de Salud del Niño.
Sólo en uno de los pacientes se demostró DUP materna parcial en el cromosoma 7 utilizando las pruebas de CMA y PCR multiplex a los padres; quien tenía fibrosis quística por una deleción completa del gen CFTR34. Otra alteración rara fue el de una paciente con un síndrome de genes contiguos (tricotiodistrofia tipo 4 /aciduria glutárica III), de herencia recesiva autosómica, donde ambos padres eran portadores recesivos de la microdeleción 35.
En el 4,7% de los casos con CNVs patogénicas por encima de 5 Mb, estos contaban con un estudio cromosómico convencional previo normal. Esto se podría deber a dos razones, la primera sería que el cariotipo no se realizó con los estándares de confiabilidad; y la segunda estaría en razón que algunas clonas con las anomalías cromosómicas tienden a desaparecer en el tiempo con el fin de corregir esos errores 36,37 y que el análisis cromosómico por micromatrices tiene una posibilidad de detectar mosaicismos con una mayor precisión.
Por otro lado, en un paciente se detectó un cromosoma marcador; sin embargo, el CMA resultó normal, lo cual se debería a que los marcadores utilizados en con esta plataforma no estarían ubicados en el cromosoma marcador, o que este sea conformado por más de un cromosoma, y la otra posibilidad es que el cromosoma marcador este conformado sólo por heterocromatina 38. Evaluando esta información, con los valores de sensibilidad y especificidad encontradas en este estudio (53,7% y 98,9%), y los valores predictivos positivo y negativo, asumiendo que el 1% de la población tiene un CNV patogénico 39; se encontró que estos valores son muy similares a los encontrados por Saldarriaga et al (2015) donde la sensibilidad fue de 62,6 % (IC 95% 40,8%-80,2%) y la especificidad de 99,9% (IC 95% 99,8%-100,0%) 40.
Doce niños eran portadores heterocigotos del síndrome de hipoacusia/infertilidad (MIM # 611102) de herencia recesiva autosómica, causado por la variante en deleción homocigota de los genes STRC y CATSPER241,42. Según la ley de Hardy-Weinberg, el número de heterocigotos es de 3 por cada 100 personas y que la incidencia de los homocigotos estaría en 22,5/100 000 habitantes el cual estaría por encima de muchas entidades genéticas.
Por otro lado, este análisis muestra que los padres tenían más de 30 años de edad, lo que es coherente con lo mostrado por estudios previos, en los que se evidencia que la edad parental es un factor de riesgo 43,44.
Dentro de las limitaciones del estudio están la no realización del CMA a los padres de los niños zon CNVs patogénicas, probablemente patogénicas o significado incierto, esta información hubiese sido importante para verificar si éstas fueron de novo o heredadas, evaluar la penetrancia, así como para la reclasificación de las CNVs de significado incierto. En ese mismo sentido, en el Perú no tenemos bases previas sobre la frecuencia de las CNVs en población en general.
El análisis cromosómico por micromatrices en niños que presentan retraso del desarrollo psicomotor, discapacidad intelectual, y síndrome malformativos; es una prueba que tiene una eficacia superior para determinar CNVs patogénicas o probablemente patogénicas a los test convencionales utilizados en nuestro medio (ej. cariotipo). Se ha observado una tasa de detección en el INSN, según la patología, entre el 47,0% y 54,5%. Estos resultados reflejan la necesidad de brindar el asesoramiento genético exacto, pues permitirá conocer el pronóstico de la entidad y realizar un manejo mucho más adecuado. En este sentido, esta información demuestra ser también superior al promedio reportado. Además, al detectar los ROH mayor a 2,56% a través del CMA, identificamos que existe una consanguinidad no declarada hasta en dos veces lo reportado en otras regiones, lo cual podría estar relacionado a situaciones de abuso sexual intrafamilar en nuestro medio. Sin embargo, existe un grupo de pacientes que no tuvieron diagnóstico etiológico, por lo que es de suma importancia implementar otras técnicas masivas como el secuenciamiento por paneles, exómico o genómico, así como una masificación de estas pruebas en otras instituciones de nuestro país.

