











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El síndrome de Guillain-Barré (SGB) es una polirradiculopatía inflamatoria aguda o subaguda poco común. Se estima que lo sufren 1 o 2/100 000 habitantes, puede afectar a personas de cualquier edad; y suele aparecer unos días o semanas después de que la persona haya tenido síntomas de infección viral respiratoria o gastrointestinal. Los criterios requeridos para el diagnóstico de un SGB son debilidad progresiva y arreflexia; y aunque, se suele solicitar exámenes del líquido cefalorraquídeo (LCR) y del electromiograma (EMG), estos hallazgos solo apoyan al diagnóstico clínico, ya que inicialmente pueden ser absolutamente normales 1.
Se han descrito cuatro variantes de SGB: polineuropatía desmielinizante inflamatoria aguda (AIDP), síndrome de Miller Fisher, neuropatía axonal sensitivo motora aguda (AMSAN), y neuropatía axonal motora aguda (AMAN). De éstas, la variante axonal motora es la de peor pronóstico, debido a que la destrucción del axón hace improbable su regeneración completa, representando el 30% a 47% de los casos en el Asia, Centro y Sudamérica 2,3.
Sin embargo, existen variantes del SGB de muy poca presentación como la debilidad distal de extremidades (DL-GBS), que es una variante regional propuesta recientemente, con la característica debilidad limitada en las extremidades distales (muñecas, manos, tobillos y dedos de los pies) y fuerza muscular conservada en las extremidades proximales (hombros, codos, caderas y /o rodillas) durante todo el curso de la enfermedad. Se estima que estas variantes representan el 7,5% de todos los casos del SGB, están generalmente asociadas a infecciones por Campylobacter jejuni, virus de Epstein Barr, citomegalovirus o Mycoplasma pneumoniae; presentan niveles normales de proteínas en el LCR, anticuerpos antigangliosidos positivos y estudios de electromiografía y neuro conducción de tipo axonal 4-7.
Estudios epidemiológicos previos muestran que las formas leves del SGB comprometen más frecuentemente a los hombres y menores de 50 años, en comparación a las formas severas del SGB, alcanzando el pico de afectación clínica (Nadir) en tiempos similares al de las personas con enfermedad más grave 8,9. El estudio de formas leves del SGB puede contribuir a conocer más del espectro de este síndrome y explorar sobre la necesidad de tratamiento con inmunoglobulina EV en estos casos.
REPORTE DE CASO
Mujer de 26 años, ingeniero industrial, soltera, agnóstica, de nivel socio económico medio, con alimentación balanceada, recibió inmunización contra influenza hace un año, no consumidora de alcohol ni tabaco. Sufre de anemia crónica de tipo ferropénica, y fue sometida a una cirugía por miomatosis uterina hace un año (2019).
Dos semanas antes del ingreso por emergencia, presenta leve déficit motor distal de la extremidad superior izquierda, inmediatamente después compromete la extremidad superior contralateral y días después afecta las extremidades inferiores distalmente; concomitantemente durante el trascurso de la enfermedad presenta diarrea que remite espontáneamente. Al examen neurológico, se encuentra despierta, orientada, sin trastornos de funciones cerebrales superiores, no compromiso de pares craneales, leve cuadriparesia distal (fuerza muscular: 4/5), sin trastorno sensitivo, sin hipo/arreflexia, no Babinsky, no trastorno cerebeloso y signo de Lasegue bilateral. En la escala de evaluación del MRC-sumscore at nadir (Medical Researh Council) 10 obtuvo una puntuación de 50/60 en general, con una distribución de 25/30 en las dos extremidades de ambos lados por separado.
Las pruebas de laboratorio mostraron: velocidad de sedimentación globular (VSG): 5 mm/h, proteína C reactiva (PCR): 0,32 mg/L (< 6 mg/L), hemoglobina (Hb): 10,7 g/dL, hematocrito (Hto): 35,5 %, VCM: 70 fL, HCM: 21,1 pg, CHCM: 30,1 g/dL, Anisocitosis 1+, Microcitosis 1+, reticulocitos: 1,8 %, leucocitos: 7,100 (neutrófilos: 65% y linfocitos: 24,1%), bandas: 0%, plaquetas: 342 000, tiempo de protrombina: 13.5 seg, índice internacional normalizado (INR): 1,13, TPT: 28,8 seg, fibrinogeno: 255 mg/dL. Por otro lado, se tomó un examen general de orina y un perfil hepático con resultados normales (Tabla 1).
Tabla 1 Valores obtenidos en estudios de laboratorio realizados en paciente con déficit motor distal.
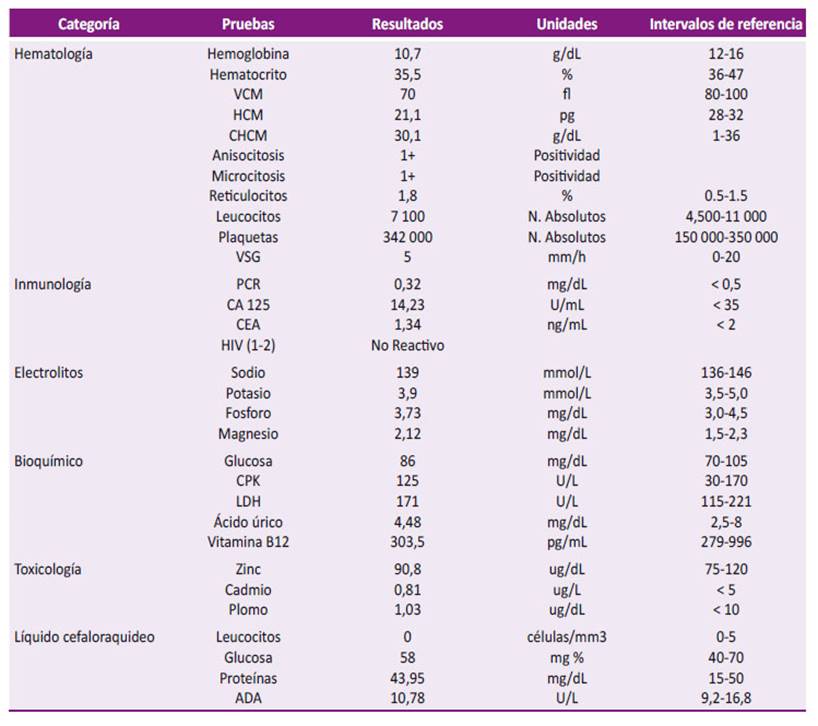
En el perfil bioquímico, se encontró glucosa: 86 mg/dL, urea y creatinina en valores normales; fosforo: 3,73 mg/dL, magnesio: 2,12 mg/dL, sodio: 139 mmol/L, potasio: 3,9 mmol/L, cloro: mEq/L; creatinfosfoquinasa (CPK): 125 U/L, deshidrogenasa láctica (DHL): 171 U/L, vitamina B12: 303,5 pg/mL, ácido úrico: 4,48 mg/dL.
Las pruebas inmunológicas mostraron: Hepatitis C (HVC): 0,036; Anti core (HBc Total): 2,25 COI, herpes virus 1 (HVS 1 IgG): 250 U/mL, herpes virus 2 (HVS 2 IgG): 96,3 U/mL, rubeola (Ac IgM y Ac IgG) 0,578 UI/mL y 146 UI/mL, Toxoplasma Gondi (Ac IgG y Ac IgM): 7,2 UI/mL y 0,027UI/mL, citomegalovirus (CMV Ac IgG y Ac Ig M): 11,6 y 0,368 ratio, e HIV (1-2): 0,416 COI (No reactivo). Los marcadores antitumorales mostraron: CA 125: 14,23 U/ mL, CEA: 1,34 ng/mL; y las pruebas toxicológicas de metales: zinc 90,8 ug/dL, cadmio 0,81 ug/L, y plomo 1,03 ug/dL. El estudio de líquido cefalorraquideo (LCR): Leucocitos: 0/mm3, proteínas 43,95 mg/dL, glucosa 58 mg%, adenosina desaminasa (ADA) 10,78 U/L. Se solicitaron pruebas para anticuerpos anti-Campylobacter jejuni y anticuerpos antigangliósidos GM1 y GD1, los cuales no pudieron realizarse por limitaciones técnicas de Laboratorio.
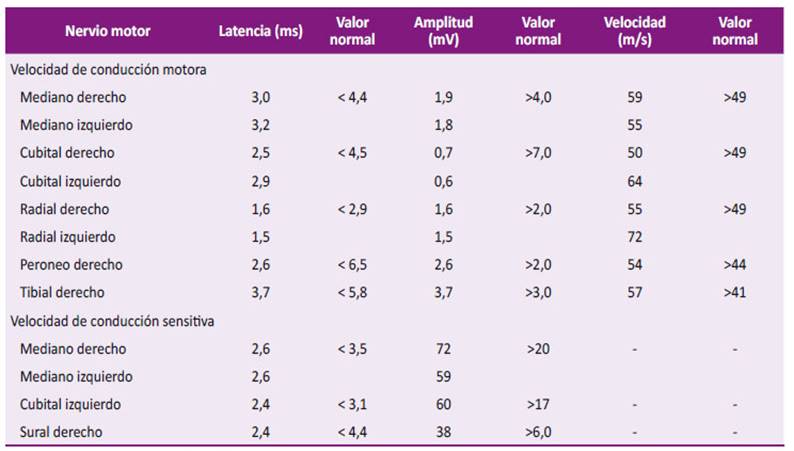
Se solicitaron estudios de velocidades de conducción al décimo día de la enfermedad, los resultados de las latencias motoras y sensitivas distales fueron normales, disminución de las amplitudes de los potenciales de acción motor de los nervios mediano, cubital, radial y peroneo de ambos lados, los potenciales de acción sensitiva normales, y las velocidades de conducción motoras y latencias de la onda F dentro de la normalidad. No se evidenciaron denervación en el estudio (Tabla 2). Se realizaron estudios de RMN cerebral y columna cervical con resultados normales.
DISCUSIÓN
El SGB está considerado como una enfermedad autoinmune, en la cual la exposición a agentes infecciosos (C. jejuni, citomegalovirus, virus de Epstein-Barr, Mycoplasma pneumoniae, entre otros) activan una respuesta inflamatoria mononucelar mediada por linfocitos T activados en contra de proteínas de mielina de las células de Schwann, lo que da lugar a la destrucción de éstas por parte de macrófagos 11. Las variedades axonales, en especial la motora aguda, presentan un proceso patológico diferente mediado por el receptor de la fracción Fc del anticuerpo dirigido contra los antígenos de los gangliósidos (GM1, GD1, GQ, GA1 entre otros) localizados en la axolema, lo que permite la invasión de los macrófagos en el nodo de Ranvier y la destrucción selectiva del axón, dejando íntegra la célula de Schwann 12,13. El caso que presentamos es una variante clínica del SBG de características clínicas leves (Nivel 2 de los Criterios de Brighton), que representa a un tercio de todos los pacientes con SGB con afectación para la marcha, de acuerdo con algunos reportes en Europa 14. A diferencia de la variante clásica del SGB, los pacientes con la forma axonal motora aguda no manifiestan alteraciones sensitivas, pero, pueden presentar hiperreflexia como manifestación inicial, lo que confunde el cuadro clínico y hace necesario descartar enfermedades de sistema nervioso central.
En el caso presentado, las características clínicas cobran gran interés debido a que el paciente comenzó concomitantemente a una infección enteral y con biparesia braquial distal de progresión descendente SIN hiporreflexia/arreflexia. Se tuvo que descartar una enfermedad desmielinizante del SNC como la esclerosis múltiple mediante resonancia magnética cerebral y de columna y así mismo se descartó una polineuropatía motora axonal de etiología toxica, nutricional o paraneoplasica con los estudios laboratoriales correspondientes por las características clínicas y los resultados de una Neuropatía Motora Axonal Aguda en la EMG. El estudio de LCR mostro acelularidad sin proteinorraquia que apunta a favor de una afección axonal más que de una afectación desmielinizante periférica, en donde el patrón clásico es la elevación de proteínas.
El fenómeno de hiperreflexia se ha descrito en el 13% de los casos con SGB, y la mayoría de los casos se presenta en pacientes con forma axonal motora aguda. Al revisar la literatura, los casos de hiperreflexia se presentan comúnmente como un fenómeno tardío hacia la segunda semana de evolución 15,16, nuestro paciente no presento hiperreflexia, pero tampoco hipo o arreflexia, los reflejos osteotendinosos estuvieron normales. La hiperreflexia asociada a este tipo de neuropatías se puede explicar por daño en el sistema nervioso central, pero más habitualmente porque la vía aferente del reflejo (sensitiva) está íntegra y es capaz de estimular las motoneuronas gamma de la médula en las etapas tempranas o tardías de la enfermedad, para estimular la motoneurona inferior, ya que esta última en estas etapas no se ha destruido del todo, o ya presenta una recuperación parcial 17.
Se decidió iniciar el tratamiento con inmunoglobulina endovenosa (IgIV): 0.4 gr/Kg/día por cinco días debido a la forma axonal aguda del SGB, por el antecedente de diarrea o posibles disautonomías frecuentes en la variante axonal. La mejoría clínica del paciente fue evidente en las extremidades inferiores, pero no así en las extremidades superiores en las que el déficit motor distal permaneció inalterable hasta el momento del alta. Si bien es cierto que el beneficio de las IgIV en las formas leves del SGB es aún desconocido, la presencia de diarrea en el cuadro clínico, ha sido relacionado como un factor de mal pronóstico en el SGB tratado con IgIV 14. Sin embargo, también se ha observado que estos pacientes presentan una mejoría significativamente mayor en el grado de discapacidad después de cuatro semanas del tratamiento con IgIV, en comparación con la plasmaféresis, como fue el caso de nuestro paciente 18.
Este caso por las características benignas y su evolución se trataría de una variante leve del SGB (DL-GBS), que difiere de las variantes normales del AMAN-SGB que generalmente son graves y de pobre pronostico, lo que justifica la posición de autores como Oshima et al., (2002), que proponen una clasificación diferente al SGB argumentando que existe un grupo de síndromes neuropáticos asociados a hiperreflexia y anticuerpos antigangliósidos diferentes al SGB 19.

