











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
En la actualidad, sabemos que las citocinas, moléculas de 30 kda, tienen una función de mensajeras encargadas de llevar información desde una célula a otra. Participan en procesos de inmunidad innata como adquirida (1, 2, 3) por lo que se ha postulado que las mismas citocinas pueden generar una respuesta inmunológica desregulada, 4 caracterizando muchas enfermedades autoinmunes que podrían constituir una de las bases fisiopatológicas de la exagerada respuesta inflamatoria que se desarrolla durante la sepsis y, más recientemente, en la infección por SARS-CoV-2.
Para que estas citocinas puedan interactuar con la célula y ejercer su trabajo, tienen que interactuar con receptores intracelulares de citocinas. En los últimos 20 años, (Esquema 1) se han generado nuevos conocimientos, es así que, se ha identificado uno de los receptores más importante en relación con la patogenia de las enfermedades inmunes, tal es el caso del receptor tipo I/II de citoquinas que usa el sistema Janus Kinasa - transducción de señales y activación de la transcripción (JAK STAT) para su funcionamiento.
Las Janus Kinasa (JAK) son un conjunto de enzimas citoplasmáticas que hacen de mensajeras facilitando la transmisión de señales desde la superficie celular al interior de esta. (6, 7). Estas enzimas fueron descritas por primera vez en 1990 6 y, hasta el momento se han descrito cuatro componentes de esta familia: JAK1, JAK2, JAK3, TyK2. 7. El primero en describirse fue el componente TyK2, posteriormente se fueron descubriendo el resto de componentes del sistema. (8,9, 10). El sistema de transducción de señales y activación de la transcripción (STAT) fue descrito en 1992 11 se han descubierto siete proteínas: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, STAT6. 12).
En la presente revisión, se hará la descripción desde la fisiología del sistema JAK/STAT, su mecanismo en ciertas enfermedades, su relación con la sepsis, la atractiva interacción que tiene la enfermedad por COVID-19 en este campo para, finalmente, poder establecer un enfoque traslacional aplicado en el enfermo crítico.
VÍA JAK-STAT: UN CAMINO POR LA FISIOLOGÍA
La vía JAK-STAT ha sido utilizada por más de 500 millones de años, siendo un camino para transmitir señales desde los receptores extracelulares al núcleo.
Para poder entender cómo funciona este sistema, es importante conocer en primer lugar su conformación. Clásicamente, en el núcleo del sistema JAK-STAT se han identificado 3 dominios proteínicos (13, 14):
Proteína tirosin-kinasa (PTK): fosforila los residuos de tirosina, escribe el código de información.
Src-homóloga 2 (SH2): se une a los residuos de fosfotirosina alrededor de la tirosina, lo que permite leer el código.
Proteína tirosin-fosfatasa (PTP): permite la desfosforilación de la fosfotirosina lo que borra el código de información.
Además de estos 3 componentes, el sistema JAK-STAT consta de otros dominios que proporcionan funciones accesorias:
Dominio FERM: consiste en tres subdominios (F1, F2, F3) que tienen una estructura similar con la ubiquitina; la función de este dominio permite mediar las interacciones proteína- proteína.
Caja SOCs: media las interacciones con los componentes de la vía de degradación proteasomal, particularmente ubiquitina- ligasas, regulando así la vida media de las proteínas. 15.
Dominio de Unión de ADN: Presente en la porción STAT que se encarga de mediar la interacción con el ADN.
Dominio espiral: involucrado en interacciones proteína- proteína.
Dominio de transactivación (TAD): es importante para el reclutamiento de cofactores y las respuestas transcripcionales. (16, 17, 18).
Toda la señal se inicia cuando las citocinas se unen a sus receptores I/II. 4. Estos receptores, se oligomerizan lo que permite la separación de las subunidades intracelulares del receptor de cinasa, activando el sistema JAK. Una vez activado, se fosforilan a sí mismos y a la porción intracelular de sus receptores, creando sitios de unión para los dominios homólogos a Src2: SH2. (19, 20, 21). Además, este paso sirve como sitio de acoplamiento para los factores de transcripción STAT.
Una vez activado los STAT, logran la translocación del núcleo donde actúan como factores de transcripción para regular la expresión génica. 4.
Podemos explicar la vía JAK STAT (figura 1 y 2) de la siguiente manera 22,23:
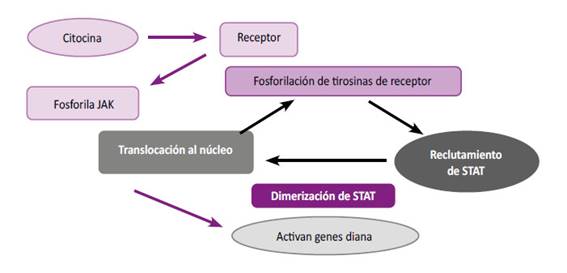
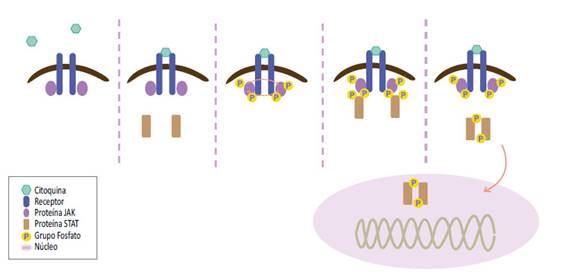
Primer paso: Unión de la citoquina a su receptor, permitiendo fosforilación de las proteínas JAK que fosforilan a los restos tirosina, las cuales reclutan a las proteínas STAT.
Segundo paso: Fosforilación de las proteínas STAT que se dimerizan y se translocan al núcleo.
Tercer paso: ya en el núcleo estas proteínas STAT activan la transcripción de los genes, que muchas veces tienen que ver con la acción del sistema inmunitario.
La vía del JAK/STAT tiene algunos reguladores negativos 12 que modulan la respuesta:
Proteína tirosina fosfatasa (PTP): facilita la desfosforilación.
Supresores de la señalización de citocina (SOCS).
Inhibidores de proteínas activadas (PIA): proteínas que pueden unirse a dímeros STAT activados y evitar que se unan al ADN.
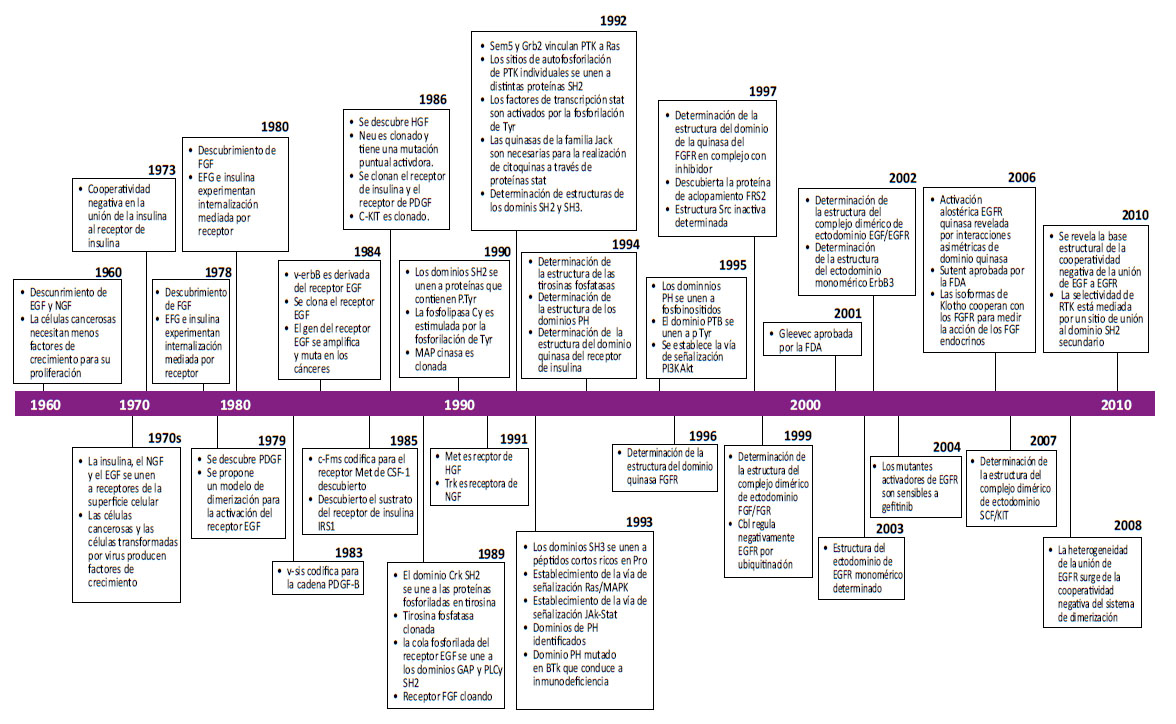
Esquema 1: Línea de tiempo de los hallazgos clave durante la historia de los recetores tirosin cinasa, con énfasis en los hallazgos y descubrimientos que produjeron el marco conceptual en el desarrollo de dicho campo y su aplicación clínica. (Adaptado de Lee et al. 1985; Libermann et al. 1985; Margolis et al. 1990; Bottaro et al. 1991; Bae et al. 2009, Schlessinger 2014)
Primer paso: Unión de la citoquina a su receptor, fosforilación de las proteínas JAK que fosforilan a los restos tirosina, las cuales reclutan a las proteínas STAT
Segundo paso: Dimerización de las proteínas STAT que se translocan al núcleo
Tercer paso: Las proteínas STAT activan la transcripción de los genes (detalle en la figura 2)
Haremos una especial mención a las SOCS, se han demostrado que inhiben este proceso por dos mecanismos: el primero es unirse a las tirosinas fosforiladas, lo que hace que compita con el reclutamiento de STAT (inhibición competitiva), permitiendo que las moléculas STAT no se unan. El segundo mecanismo es la inhibición que se establece sobre las moléculas JAK, lo que inhibe la fosforilación y el bloqueo de STAT al componente residual de tirosina fosforilada. (12, 24, 25).
IMPLICACIONES DE LA VÍA JAK STAT EN LA INFLAMACIÓN Y SU RELACIÓN CON ENFERMEDADES AUTOINMUNES
Se ha demostrado que tanto el factor de crecimiento hematopoyético, incluido eritropoyetina, trombopoyetina, dan su señal a través del JAK 2. Por lo tanto, una mutación de este se ha relacionado con policitemia vera, trombocitopenia esencial y mielofibrosis. (26, 27, 28, 29, 30, 31). Otra de las alteraciones importantes son las mutaciones y pérdida de función a nivel del JAK subtipo TYK2, donde el cuerpo, es más susceptible a infecciones de tipo viral debido a la pobre respuesta al interferón. (27, 28, 32). Una pérdida en la función del JAK 3 causa una inmunodeficiencia severa, afectando las acciones de células T y natural killers con sus consecuencias sobre la respuesta inmune. Llama la atención que pacientes con inmunodeficiencia combinada grave autosómica recesiva, asociada a cambios en el receptor JAK 3, no sean afectados por las manifestaciones extra inmunitarias de la enfermedad, lo que sugiere que el bloqueo del JAK 3 es una posible terapia inmunosupresora. (33, 34).
Así mismo, mutaciones en la proteína STAT, permite que el paciente padezca de infecciones por micobacterias (35, 36). Mutaciones en la proteína STAT2, podrían desencadenar infecciones de tipo viral, generando un síndrome parecido a la sepsis después de haber recibido una vacuna con virus atenuados. 37. Mutaciones en el STAT 3 se han relacionado con enfermedades autoinmunes de inicio temprano, tales como diabetes neonatal y enfermedad autoinmune linfoproliferativa. (38, 39, 40).
Dentro de la constelación de enfermedades asociadas a cambios en el sistema JAK/STAT se han filiado a la artritis reumatoide 41, psoriasis (42, 43) y enfermedad intestinal (44, 45), por lo que, al final de los años 90, la FDA aprobó fármacos llamados ‘Jakinibs’ para bloquear esta vía (46, 47).
RUTA DE SEÑALIZACIÓN JAK-STAT ASOCIADA A LA SEPSIS
La sepsis, como inductor de fallo multiorgánico, tiene un papel fundamental en la mortalidad dependiendo de la severidad con que se presente cada una de sus fases. El síndrome de respuesta inflamatoria sistémica (SIRS) seguido de un estadio de inmunosupresión inducida por sepsis o respuesta antiinflamatoria compensatoria (CARS), tienen como finalidad equilibrar la secreción de citocinas pro y antiinflamatorias (48, 49). Tanto SIRS como CARS, introducen a la Janus Kinasa o proteína JAK además de transductores y activadores de señal de transcripción o proteína STAT. (4, 26).
En cuanto a la función de las proteínas JAK-STAT en la sepsis, existen múltiples mecanismos por los cuales se puede desencadenar una respuesta desregulada del huésped 50. La SIRS está mediada principalmente por STAT1 51 y STAT4 52, mientras que el CARS predominan STAT3 (53) y STAT6, con la participación de JAK1-2 y tirosina quinasa 2 para ambos casos. (12, 54).
Asociado a la proliferación celular y apoptosis, las proteínas JAK y STAT se han visto incluidas en el proceso de hematopoyesis de emergencia y disfunción orgánica que surge en un paciente séptico, lo cual es indicativo de que el dirigir la terapéutica a la vía de JAK/STAT pudiera significar cambios clínicos a favor del paciente con sepsis. (54, 55).
Las proteínas JAK y STAT desempeñan funciones muy relevantes durante la sepsis, a través de la defensa del huésped contra los distintos patógenos y regulación de la inmunidad. Recalcando la importancia que tiene el reconocimiento de patrones moleculares asociados a patógenos (PAMP) por los receptores de reconocimiento de patógenos (PRR), que activan el sistema inmunitario innato durante la sepsis, los cuales van a necesitar de dichas vías de señalización para cumplir su objetivo. 11.
Estas vías incluyen a los receptores del factor estimulante de colonias de granulocitos (G-CSF), proteína inflamatoria de macrófagos tipo 2 (también llamada ligando 2 de quimiocina CXC, CXCL2), citocinas de tipo I, incluidas las interleucinas (IL-4, IL-6, IL -10, IL-12 e IL-13) y citocinas de tipo II, incluidos los interferones (IFN-α, IFN-β e IFN-γ). (56, 57, 58).
Por lo tanto, JAK y STAT están implicadas en la señalización de citocinas tanto proinflamatorias (IFN-γ, IL-12 e IL-27) como antiinflamatorias (IL-4, IL-10 e IL-13), lo que, a su vez, revela la capacidad de varios miembros de la familia STAT para regular diferencialmente el equilibrio de las células T helper (Th) 1 /Th2, que desempeñan un papel fundamental dentro de la sepsis. (13, 56). En la Tabla 1 se detalla el rol que desempeña cada uno de los miembros de la familia JAK y STAT en la respuesta del huésped contra la sepsis bacteriana 55.
Tabla 1 Funciones respectivas de los miembros de la familia JAK y STAT en la respuesta del huésped contra la sepsis bacteriana.
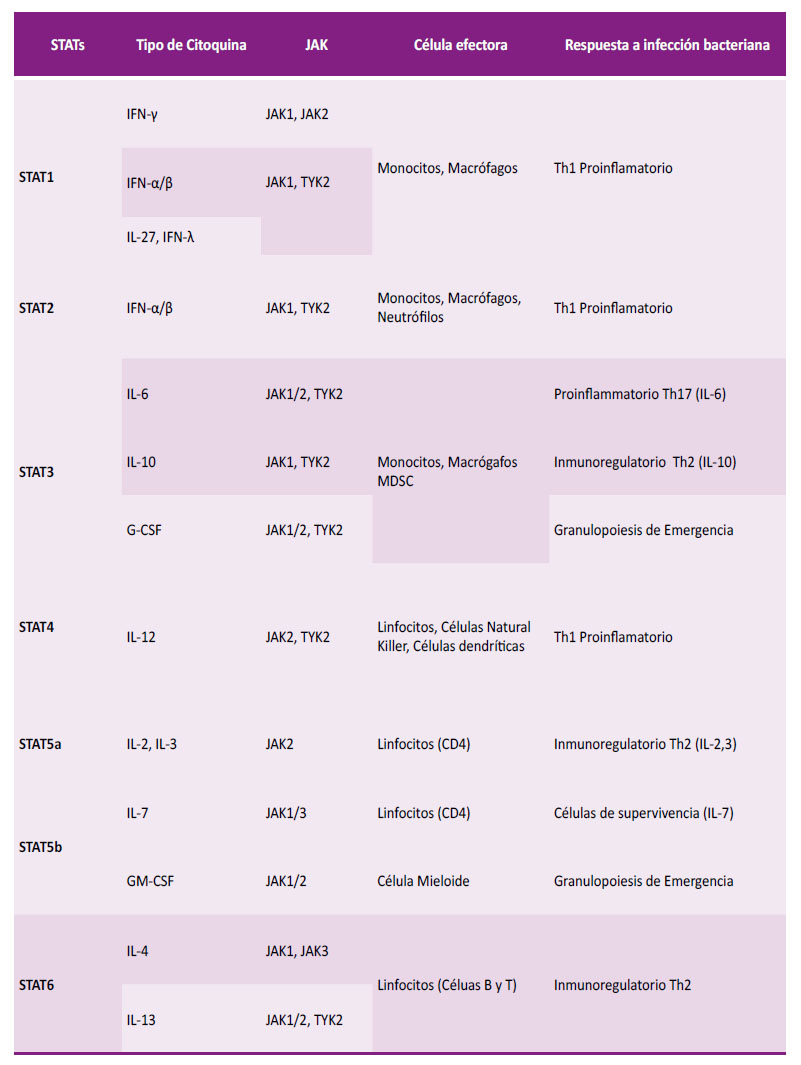
JAK: Janus-Kinasa, STAT: transducción de señales y activación de la transcripción IFN: interferón IL: interleuquina G-CSF: factor estimulante de colonias de granulocitos GM-CSF: factor estimulante de colonias de granulocitos y monocitos
La presencia de STAT en los distintos tipos de receptores, permite una señalización parcialmente restringida a cada tipo de célula en respuesta a las citocinas. De esta manera, la activación de macrófagos está regulada principalmente por citocinas que utilizan STAT1, que participan en la expresión inducible de óxido nítrico sintasa (iNOS) y en las vías de señalización de STAT3. Por otro lado, los neutrófilos requieren STAT2 que mejoran la actividad de superóxido dismutasa. Los linfocitos, a su vez, están regulados principalmente por STAT4 y STAT6 para las respuestas proinflamatorias Th1 e inmunorreguladoras Th2, respectivamente. Las proteínas STAT4 también son requeridas usualmente para respuestas mediadas por IL-12 de células Natural Killer y células dendríticas. Aunque las vías JAK-STAT son de gran importancia para la inmunidad innata, tienen su contraparte al participar en la hiperinflamación e inmunosupresión que siguen a la sepsis. (figura 3).
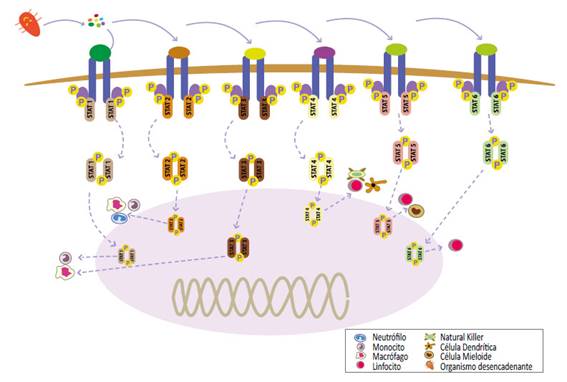
Figura 3 Participación de las vías JAK-STAT en las cascadas de citokinas que producen hiperinflamación e inmunosupresión debido a la sepsis.
Al realizar un análisis sobre cómo la vía de estas proteínas inducen un proceso hiperinflamatorio y, a su vez, tiende a incrementar la mortalidad en los pacientes sépticos, podemos ver que son estas mismas vías de señalización JAK/STAT las que inducen la producción de moléculas proinflamatorias que requieren especialmente STAT1, STAT4, TYK2 y JAK2, así como en la mielopoiesis de emergencia, la cual no es otra cosa, que la producción rápida y masiva de células inmunitarias, necesaria para la lucha contra los patógenos bacterianos, siendo un proceso fundamental que acompaña a la respuesta inmune durante la sepsis. 4. STAT3 es fosforilado por JAK1/2 y TYK2 durante la mielopoiesis de emergencia y juega un papel importante en este proceso, debido a que la proteína STAT 3 fosforilada regula de forma ascendente la transcripción de los genes que codifican MIP-2 (CXCL2) y su receptor (receptor de quimiocina del C-X-C, CXCR2). (4, 59, 60, 62).
Como es bien conocido, el estado hiperinflamatorio resultante mediado por JAK-STAT durante el choque séptico, contribuye a la mortalidad por daño tisular y síndrome de fallo multiorgánico, lo que obliga al huésped a generar mecanismos de compensación antinflamatorios. Este estado, conocido como CARS, involucra predominantemente a STAT3 y STAT6, recalcando que es este el mecanismo de regulación que inicia desde el comienzo del choque séptico debido a la necesidad de proteger las células y a los tejidos de una respuesta inmune inflamatoria excesiva durante la enfermedad. 62.
Existen varios receptores de citocinas antiinflamatorias como la IL-10 que regula negativamente los genes proinflamatorios inducidos por IFN suprimiendo la fosforilación de STAT1 e iniciando la respuesta antiinflamatoria Th2 mediada por STAT3 que, en su estado activo, limita la producción de TNF e IL-6. 55.
Los STAT activados inducen la transcripción de genes SOCS, que forman parte de un mecanismo de control del sistema JAK STAT, facilitando su degradación mediada por ubiquitina, previniendo su fosforilación y, por ende, el ingreso al núcleo para producir el estado hiperinflamatorio. Así, el SOCS3 contribuye al control de la hiperinflamación inhibiendo STAT1, STAT4 y STAT5, mientras que SOCS1 interactúa con TYK2 y STAT1 63.
Teniendo en cuenta que la sepsis se caracteriza por un cuadro de disfunción endotelial que causa cual una parálisis vasomotora, coagulopatía de consumo, fuga capilar y daño multiorgánico, la proteína JAK3 se expresa en células endoteliales vasculares, especialmente cuando es estimulado por LPS, TNF, IL-1β e IFN-γ. (64). A través de la fosforilación de STAT3 y la activación de NF- κB, la proteína JAK3 impulsa la sobreexpresión de las moléculas de adhesión endotelial y molécula de adhesión intercelular (ICAM) -1, molécula de adhesión de células vasculares (VCAM) -1 y molécula de adhesión de células endoteliales plaquetarias (PECAM) - 1, lo que permite la unión de leucocitos y plaquetas al endotelio y su transmigración a los tejidos periféricos. (56, 64)
Mientras que la expresión de PECAM-1 mediada por STAT3 mantiene la integridad endotelial, la ICAM1 promueve la transmigración de leucocitos mediada por STAT3 y aumenta la permeabilidad microvascular general, que es el inicio de la fuga capilar inducida por sepsis. En contraposición, STAT2 controla la salida celular excesiva mediada por ICAM-1, lo cual pudiera contribuir a una mayor letalidad por un mecanismo centrado en la disfunción endotelial incrementada, en conjunto con la sobreexpresión de ICAM-1 y la transmigración celular que conlleva a ello. 65.
La respuesta proinflamatoria STAT1 y STAT2 (Th1) está mediada por el factor génico estimulado por interferón 3 (ISGF-3), que es un complejo de factor de transcripción heterotrimérico que asocia STAT1, STAT2 y el factor regulador de interferón 9 (IRF- 9). STAT4 a su vez, ejerce efectos patógenos al promover las respuestas inmunitarias Th1 involucradas en fallas renales y hepáticas agudas e inflamación pulmonar que conduce inclusive a desarrollar el típico síndrome de dificultad respiratoria aguda (SDRA). (64, 65)
Por otra parte, STAT6 media la respuesta Th2 y actúa de dos formas diferentes:
Restringiendo directamente la inflamación hepática
Comprometiendo el aclaramiento bacteriano mediado por Th1 y exacerbando la lesión renal aguda y la inflamación pulmonar. 44.
La vía JAK3/STAT3 también se asocia con insuficiencia miocárdica directa e indirectamente al promover la disfunción endotelial, la cual estará mediada por STAT3 con importante fuga capilar, vasoplejía y coagulopatía intravascular diseminada (CID), lo que, a su vez, exacerba la hipoxia tisular observada en el choque séptico, considerando que este tipo de lesiones tisulares son responsables de la generación de DAMP, que son los que van a reforzar la respuesta proinflamatoria. (22, 44, 65). En un análisis global sobre el mecanismo fisiopatológico de la sepsis, todo empieza con el ingreso del microorganismo y sus demás componentes, 64 los cuales van a desatar en el individuo cuatro respuestas principales: (figura 4 ).
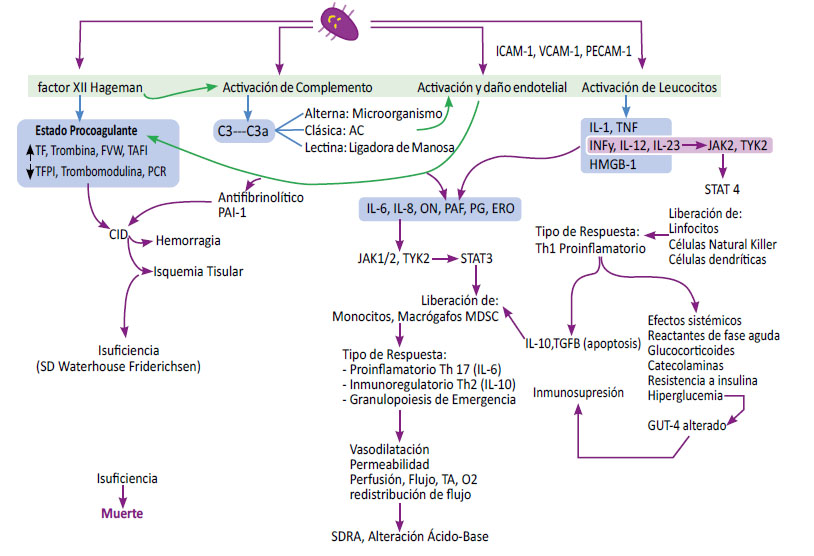
Figura 4 Participación de las vías JAK-STAT en la activación y reclutamiento de diferentes líneas celulares de leucocitos, mediados por la producción inicial de ICAM-1, VCAM-1, y PECAM-1.
La primera, aunque no es de forma cíclica ni en orden jerárquico, tiene que ver con la activación del factor VII o también llamado factor Hageman, lo cual va a activar la vía intrínseca de coagulación, con un consecuente estado procoagulante donde se generará aumento del factor tisular, trombina, exposición del factor Von Willebrand, TAFI o factor inhibidor de la fibrinolisis con disminución de sus contrapartes, fundamentalmente TFPI, trombomodulina y proteína C, estableciendo un estado procoagulante. 66.
Otro mecanismo, es mediante la activación del complemento, con sus tres principales vías: la vía alterna, en la cual interviene directamente el microorganismo; la vía clásica a través de anticuerpos; la vía de las lectinas mediante la lectina ligadora de manosas 67. Un tercer mecanismo se da mediante la activación y daño endotelial y, finalmente, un cuarto mecanismo es por medio de la activación directa de leucocitos, estos dos últimos, mediados por la producción inicial de ICAM-1, VCAM-1, PECAM-1, que es fundamentalmente donde influirá la vía JAK-STAT. (64, 67).
A su vez, el complemento también va a permitir la activación y lesión endotelial que, de igual manera, conlleva un estado procoagulante con mayor liberación del factor Von Willebrand asociado a otras dos grandes respuestas. Una de ellas es un estado antifibrinolítico, con incremento de PAI-1 y, a la par, se genera la liberación de IL-6, IL-8, óxido nítrico y factor activador de plaquetas (PAF), prostaglandinas y especies reactivas de oxígeno (ERO) que, a su vez, permitirán que la proteína JAK-1 y JAK-2, generen fosforilación de STAT3, teniendo como desenlace la liberación de monocitos y macrófagos MDSC con 3 tipos de respuesta fundamentalmente proinflamatoria, secundario a Th17, mediado por IL-6, Inmunoregulatoria Th2, debido a IL-10 y Granulopoiesis de emergencia. (55, 67).
A su vez, la activación de los leucocitos, inicialmente en una fase temprana, van a liberar IL-1, TNF y en estados más avanzados y conforme va progresando la enfermedad, liberará interferón gamma, IL-12, IL-23 y, en una fase tardía, HMGB-1 conocida como proteína de alta movilidad. Mediante la proteína JAK2 y TYK-2, forsforilarán a STAT4, con la consecuente liberación de linfocitos, células NK y células dentríticas, con una respuesta de tipo Th1 proinflamatoria, generando la liberación de interleuquinas reguladoras como la IL-10. (55, 56, 68, 69).
Estas interleuquinas también desencadenan un estado de vasodilatación, con aumento de permeabilidad, generando clínicamente edema, disminución de la perfusión y con ello menor flujo sanguíneo, hipotensión arterial e hipoxemia, con redistribución de flujo, disminuyendo sobre todo en áreas no vitales para distribuirlo a órganos blancos. 67,68.
Dentro de los efectos sistémicos tenemos principalmente la alteración de la frecuencia cardíaca, que inicialmente responde con un aumento y progresivamente va a ir disminuyendo, se asocia en una fase temprana episodios de fiebre, estado de caquexia, aumento de reactantes de fase aguda, liberación de glucocorticoides y catecolaminas, resistencia a insulina y por ende hiperglicemia asociado a una alteración de la GLUT-4. Estos efectos sistémicos, en conjunto con las interleuquinas reguladoras, llevarán al individuo a un estado de inmunosupresión. Esta alteración afecta a múltiples órganos, apareciendo compromiso pulmonar con el concomitante síndrome de distrés respiratorio, alteración del medio interno e hiperlactatemia tipo A que, inclusive, compromete la funcionalidad renal por hipoflujo sanguíneo en una fase tardía. 68.
Conforme exista mayor progresión de la enfermedad, esta situación conducirá a un estado de insuficiencia multiorgánica, por lo tanto, si no existe una adecuada intervención, llevará al individuo hacia la muerte. 68.
ENFOQUE TRASLACIONAL Y POSIBLES ESTRATEGIAS TERAPÉUTICAS DIRIGIDAS A LA VÍA JAK/STAT EN SEPSIS
La evidencia actual sugiere que la proteína STAT2 y STAT3 controlan la expresión de moléculas de adhesión endotelial, iniciando su disfunción durante la sepsis progresando en fases tardías hacia la vasoplejia, coagulopatía y fallo multiorgánico. 35.
Estudios recientes sobre el desarrollo de mielopoyesis emergente, un proceso fundamental que acompaña a la respuesta inmune durante la sepsis, demuestran que la proteína STAT3 controla la expresión de los genes C/EBPβ y Fanconi C (FANCC), los cuales son factores importantes en el proceso de inmunoparálisis; por lo tanto, la inhibición de la vía de señalización JAK/STAT podría reducir el fallo multiorgánico y la inmnosupresión. 4,59. Todo esto, debido a las recientes y exitosas terapias realizadas en pacientes oncológicos y hematológicos, brindando nuevas oportunidades para tratar a los pacientes con sepsis, sin embargo, muchas de estas estrategias terapéuticas aún están en fase de experimentación 55.
Como se analizó previamente, la sepsis es una enfermedad progresiva que involucra muchos tejidos y tipos de células, iniciada por una respuesta hiperinflamatoria seguida por el agotamiento de células inmunes. Ambas son responsables de la tasa de mortalidad elevada por esta condición.
Es importante destacar que la característica común entre otros tipos de células y sus disfunciones inmunitarias es su requerimiento de la vía JAK/STAT. Esto nos ha llevado a considerar el interés potencial de los inhibidores de JAK para modular la sepsis utilizando terapias orientadas al tiempo y a los órganos.
Los inhibidores de JAK, también llamados ‘jakinibs’, son una nueva clase de fármacos que han establecido su mayor utilidad en los últimos años. 44. Estas moléculas inhiben una o más proteínas JAK, evitando así la fosforilación y activación de las proteínas STAT correspondientes. Entre ellos, los más estudiados en la actualidad son el ruxolitinib, tofacitinib y baricitinib. El ruxolitinib, que fue el primer inhibidor no selectivo de JAK1 Y JAK2 aprobado, se desarrolló para el tratamiento de las neoplasias mieloproliferativas. Además, en estudio en ratones se observó que el Ruxolitinib podría mejorar el pronóstico de la sepsis por Candida. Los resultados obtenidos con este fármaco demostraron que al administrar una dosis de 6.25 mg/kg/día un día después del inicio de la infección mejora la supervivencia, por lo cual, la hiperinflamación se autoregula sin aumentar la carga de patógenos. 44.
Diferentes estudios experimentales han demostrado la eficacia de los inhibidores de JAK2 y confirman que el mejor momento de su administración es 24 horas después del inicio de la sepsis al disminuir la expresión de mediadores proinflamatorios como TNF, IL-6 y HMGB-1. 46.
Durante la fase aguda de la sepsis (fase hiperinflamatoria), utilizar los inhibidores de JAK/STAT puede resultar complicado. Aunque el ruxolitinib es capaz de mejorar la supervivencia durante la sepsis por Candida en ratones, se observó un riesgo de infección del 2.6 % en seres humanos. (44). Como se mencionó anteriormente, las dosis de ruxolitinib y el momento de administración influyen en gran medida en el estado inmunitario, la eliminación del patógeno y el riesgo de supervivencia. Además, se describe un síndrome de abstinencia cuando los pacientes interrumpen el tratamiento con ruxolitinib, el cual puede provocar una tormenta de citocinas perjudicando el pronóstico en la sepsis. Es por esto que, la administración sistémica de estos fármacos, es un arma de doble filo, en la que el tiempo real de inicio de la infección puede ser desconocido con mayor variabilidad del patógeno ofensor, de su carga inicial y de la respuesta inmune. Tales efectos inmunosupresores hacen que su administración sistémica sea riesgosa, lo que nos lleva a sugerir estrategias alternativas.
Estas estrategias se basan en una encapsulación de nanopartículas de moléculas terapéuticas. Es técnicamente posible y se han desarrollado varias opciones. Las nanopartículas tienen un ligando que permite dirigirse a células o tejidos específicos que van desde un órgano determinado hasta subtipos de células inmunitarias como macrófagos, células B o células T. Su objetivo principal es alcanzar un tejido altamente implicado en la fisiopatología del choque séptico, como el endotelio vascular en donde existe disfunción de las proteínas JAK3 Y STAT3 con repercusiones en el tejido miocárdico. Esta acción permite integrar un anti-STAT3 o anti-JAK3 (decernotinib) para reducir la disfunción endotelial. Además, otro objetivo sería alcanzar un tipo de célula inmunitaria específica involucrada en la hiperinflamación como los macrófagos. 59.
No menos importante, el impacto de la pandemia por la COVID-19 en el sistema de salud ha sido devastador en muchos países y existe una necesidad clínica insatisfecha de tratamientos o intervenciones terapéuticas eficaces (68, 69). Con base en lo anterior, se están desarrollando más de 10 ensayos clínicos randomizados incluyendo el uso de ruxolitinib para tratar diferentes estadios de la infección por SARS-CoV-2, los cuales prometen resultados alentadores teniendo en cuenta el perfil de seguridad del fármaco y su beneficio sobre la interleuquina 6 además de citocinas proinflamatorias y antiinflamatorias que inducen una modulación inmunitaria global. La corta vida media del fármaco (4-6h) ofrece una ventaja significativa sobre cualquier anti-IL6, ya que cualquier posible daño debido a la inmunosupresión debería revertirse fácilmente después de la interrupción del fármaco. Melatiadis et al 54 propusieron que el fármaco ruxolitinib tendría un efecto benéfico en pacientes con diagnóstico de COVID-19, bloqueando la IL-6 entre otras citocinas inflamatorias, generando una respuesta inmunomoduladora que regule los genes controlados por interferón. Sin embargo, existen algunos inconvenientes teóricos que pueden tener importantes implicaciones clínicas. El proceso de señalización JAK/STAT regula positivamente los genes controlados por interferón, una vía que con frecuencia es inhibida por productos virales, lo que da como resultado un aumento de virulencia. Por lo tanto, dicha inhibición podría facilitar la replicación viral, así como la reactivación del SARS-CoV-2 (si esto se confirma) u otros virus latentes como los virus del herpes simple o del herpes zóster 59.
Finalmente, al entender la urgente necesidad de esquemas terapéuticos para las manifestaciones clínicas de la sepsis, consideramos que la administración de agentes inmunosupresores debería implementarse solo en el contexto de ensayos clínicos aleatorizados controlados que establezcan el momento y la dosis adecuada de su administración indicando sus potenciales efectos adversos previa a su comercialización.
Perfil histórico del desarrollo del conocimiento de las vías de comunicación y señalización intracelular.
Los receptores cinasa y en general las cinasas son enzimas que transfieren grupos fosfato a sus sustratos. La importancia fisiológica de este grupo de receptores es muy amplia, pues son los responsables de mediar las respuestas a hormonas como la insulina y otros factores de crecimiento celular como el EGF (epidermal growth factor) y el TGF- (transforming growth factor-beta). Las cinasas de proteínas se clasifican de acuerdo a su especificidad por el sustrato: una clase fosforila residuos de tirosina y otra clase fosforila residuos de serina y treonina 71.
Las tirosin-cinasas, son un tipo de moléculas que tienen como función, mediar la señalización celular, estimulando de manera particular su proliferación, angiogénesis, invasión, metástasis y autorregulación. 72 De ese modo, los inhibidores de las tirosin- cinasas inhibiendo estas moléculas e interrumpiendo las vías implicadas en la proliferación celular y en la angiogénesis de las células tumorales, partiendo de ahí su relevancia clínica, ya que actualmente se las usa como terapia aprobada para el tratamiento del cáncer diferenciado y pobremente diferenciado, cuando este es resistente a yodo radioactivo y se encuentra en fase de progresión 72.
Durante la década de los 60, investigadores en Filadelfia encontraron alteraciones cromosómicas dentro del grupo G, denominándolo Cromosoma Filadelfia (Ph), siendo una de las primeras anormalidades genéticas relacionadas con cáncer, más tarde en 1973, se demostró que el cromosoma Ph era el resultado de una translocación equilibrada entre los cromosomas 9 y 22, y a partir del año 1986 cuando se establece que esta trasposición resulta en la alteración de un gen que codifica una proteína- tirosina quinasa no receptora c-ABL1, inmerso dentro de una pequeña región del cromosoma 22 a la que se le denominó región de clúster del punto de interrupción o bcr (break, cluster region). 73,74 El estudio posterior de este locus demostró que la región bcr estaba en el medio de un gran gen codificador de proteínas, principalmente los exones 12 a 16, de la región del grupo de punto de corte principal, denominados M-BCR, dando como resultado una proteína de fusión de 210 kilodaltons designada p210 BCR-ABL1 o p210 BCR-ABL, a partir de lo cual se continuó en el análisis de inhibidores de tirocin cinasa para empleo terapéutico en patologías neoplásicas 72,73
Los receptores con actividad de cinasa de tirosina se caracterizan por tener una topología que dispone que el dominio de unión al ligando esté separado del dominio cinasa por la membrana plasmática. En ausencia del ligando estos receptores están en estado monomérico y al juntarse a su ligando se induce su homodimerización. Esta dimerización permite la transmisión de un cambio conformacional del dominio extracelular al dominio citoplásmico que activa el dominio catalítico y produce la autotransfosforilación de los receptores, y de esta forma la
cinasa de un receptor fosforila residuos del otro receptor, siendo la causa de una reacción en cadena que direcciona a múltiples blancos intercelulares 75.
La función principal de los sitios autofosforilados por estos receptores, está encaminada en servir como punto de anclaje de aquellas proteínas efectoras como la fosfolipasa Cγ o la cinasa del fosfatidilnositol-4,5, bisfosfato, mismas que a su vez tienen la capacidad de ser fosforiladas por la cinasa del receptor y así propagar intracelularmente la señal del ligando. Una función adicional transductora de estas cinasas fosforiladas es la de fosforilar sustratos adicionales que inician otras vías de señalamiento, como en el caso del IRS-1 sustrato de la cinasa del receptor de insulina entre otros. 75,76
Debido a que el campo actual de los receptores Tirosin cinasa (RTK) y sus vías de señalización está cubierto por una gran cantidad de literatura que abarca más de medio siglo, nos centramos en los descubrimientos fundamentales realizados desde antes de que se descubriera la fosforilación de tirosina, haciendo hincapié en los primeros hallazgos clave, que proporcionaron marcos conceptuales para abordar las cuestiones de cómo se activan los RTK y cómo regulan las vías de señalización intracelular, resumiéndolos en el esquema 1. 76
Características de señalización en vías down stream, up stream
La red de comunicación intracelular está formada por varias vías de señalización, donde las proteínas cinasas participan en base al objetivo de la señalización y la subpoblación celular en la que se están inmersos. De esta manera, para lograr una adecuada comprensión sobre el proceso o cascada de señalización, tenemos los términos “up-stream” o corriente arriba y “down- stream” o corriente abajo, que hace analogía del flujo de una corriente de agua para indicar la dirección y la posición de las proteínas dentro de la cascada de señalización intracelular. 77
Es así como al referirnos a una acción o vía up-stream, hacemos referencia a que se genera en dirección a la membrana celular como el reconocimiento y la unión de los receptores celulares. De otro modo, al mencionar la vía down-stream significa que ocurre en dirección al núcleo celular como la transcripción génica. Por lo tanto, al mencionar que una tirosin cinasa como la JAK-3, todo aquello que la activa, como las citocinas que se unen al receptor de la cadena común, está por encima de ella en la cascada de señalización y se la cataloga como up-stream, y todo aquello que la enzima en sí misma activa, como por ejemplo la activación y el desarrollo del linfocito T y natural killer (NK) y su homeostasis, está por debajo de ella en la cascada de señalización siendo de tipo down-stream, y en este último caso incluye la regulación del crecimiento celular, la proliferación celular , la diferenciación celular y la apoptosis. 78,79
Rol como factor activador y vinculación con modelos de afección oncológica hematológica.
En la última década, se ha implementado en la práctica clínica los inhibidores tirosin quinasa como coadyuvante en el manejo de patologías oncológicas, sobre todo de tipo hematológicas, debido a que las células de leucemia mieloide crónica contienen un gen anormal, BCR-ABL, que no se encuentra en las células normales, el cual produce una proteína BCRABL, que es la causante de que las células anómalas crezcan y se reproduzcan descontroladamente, desencadenándose las cascadas mieloproliferativas. BCR-ABL es un tipo de proteína tirosina cinasa, por lo tanto, los inhibidores de la tirosina cinasa que se dirigen a la proteína BCR-ABL son el tratamiento convencional de este tipo de patologías, ya que bloquean la acción de dichas proteínas quinasas 80.
Tras comprobar su eficacia en enfermos de leucemia mieloide crónica de larga evolución que eran resistentes al tratamiento inmunomodulador con interferón o con mala tolerancia a dicho tratamiento, se llevó a cabo el estudio IRIS, en el que se probó lo que para ese momento era una molécula innovadora llamada Imatinib, capaz de bloquear la actividad desordenada de fosforilación de residuos de tirosina de segundos mensajeros que inducen procesos de proliferación tumoral en pacientes con la translocación BCR-ABL, llegó a mejorar la terapéutica de los pacientes con leucemia mieloide crónica, y permitió mejorar el entendimiento de los mecanismos de señalización, haciendo referencia a vías complejas a nivel celular, que permiten establecer otras vías predilectas para activar mecanismos de proliferación, además de vías secundarias y vías de escape a la inhibición de una molécula bloqueadora de la actividad de fosforilación de segundos mensajeros. 81,82
Este fue un estudio a nivel internacional con una extensa muestra de pacientes y que tenía como objetivo analizar la eficacia y seguridad a largo plazo de los inhibidores de tirosin quinasa BCR -ABL en el tratamiento de los pacientes con leucemia mieloide crónica, incluyéndose enfermos con LMC de 177 centros de 16 países desde el año 2000 a 2012, con edades comprendidas entre 18 -70 años, con resultados que evidenciaron una eficacia muy superior al tratamiento inmunodulador con menos efectos secundarios, tanto así que en el 90% de los casos se consiguieron respuestas citogenéticas completas, con la desaparición del cromosoma Ph de la medula ósea y en un pequeño porcentaje también se consiguió alcanzar la respuesta molecular completa, con la desaparición del gen BCR-ABL de sangre y médula. 81
A partir de ahí los inhibidores tirosin quinasa cobraron protagonismo en la práctica clínica sobresaliendo ante inmunomoduladores e inclusive por sobre el trasplante de progenitores hematopoyéticos, con respuestas muy favorables. Se destacan otros inhibidores validados para las patologías hemato-oncológicas, incluyendo el ya mencionado Imatinib, además de Dasatinib, Nilotinib, Bosutinib, Ponatinib, Asciminib.(83
Genoma Humano
A lo largo de la historia del ser humano se han documentado varias enfermedades que se dan entre familiares, pero hasta 1980 es donde la primera mutación de gen fue declarada como responsable de una patología 84,
Se define al genoma como la colección completa de ácido desoxirribonucléico (ADN) que contiene las instrucciones genéticas para desarrollar y dirigir las actividades de todo el organismo, siendo este un código de barras que cifra la información; para el 14 de abril del 2003 el Instituto Nacional de Investigación del Genoma Humano (NHGRI), el Departamento de Energía (DOE) y sus socios del Consorcio Internacional para la Secuenciación del Genoma Humano anuncian que se ha culminado con éxito el proyecto de la secuencia del genoma humano 85.
Como se mencionó previamente existe una gran relación entre defectos genéticos y enfermedades que aquejan al ser humano, es así como se ha ligado alteraciones en el sistema JAK STAT a varios procesos patológicos, los mecanismos por los cuales los STAT influyen en la transcripción génica se han esclarecido utilizando una combinación de enfoques genéticos 86,87, alteraciones en la porción JAK/STAT han demostrado que los pacientes se hacen más susceptibles a infecciones micóticas, virales, inmunodeficiencias primarias. Se ha demostrado que los STAT se unen a decenas de miles de sitios en el genoma y regulan la transcripción de miles de genes que codifican proteínas, junto con microARN y ARN largos no codificantes. Varios estudios de asociación del genoma completo han implicado la vía JAK STAT en enfermedades comunes: polimorfismos de STAT1 se han asociado con un mayor riesgo de malignidad 88 y los polimorfismos de STAT3 están asociados con la enfermedad de Crohn y la psoriasis 89.
CONCLUSIONES
La vía de señalización JAK/STAT es la principal ruta que controla la producción de citocinas en la sepsis, afectando las respuestas antiinflamatorias y proinflamatorias que desencadenan finalmente en fallo multiorgánico. Esta ruta se ha relacionado con la liberación de múltiples citocinas y mediadores inflamatorios como IL-6, IL-10, iNOS, HMGB1, entre otros, los cuales participan ampliamente en la respuesta inmune durante la sepsis. Al mismo tiempo, la vía JAK/STAT está involucrada en el choque de endotoxinas, daño orgánico y permeabilidad vascular del músculo miocárdico.
Por lo tanto, sugerimos que la vía JAK/STAT puede guiar hacia un excelente objetivo terapéutico en la sepsis. Futuras investigaciones deben ser realizadas para establecer potenciales efectos inhibidores en esta vía.

